- Home
- News & Updates
- biomodal’s duet multiomics solution +modC data now available on Illumina BaseSpace™ Sequence Hub
-
BaseSpace™ Sequence Hub
-
Publications
-
News
- 03/19/2024
biomodal’s duet multiomics solution +modC data now available on Illumina BaseSpace™ Sequence Hub
-
Sophie Wehrkamp-Richter
Discover the combinatorial power of genetics and epigenetics at single base resolution. We are delighted to present BaseSpace Sequence Hub data on the NovaSeq™ 6000 S4 flowcell for biomodal’s duet multiomics solution +modC.
Workflow
Genetics and epigenetics both offer important layers of information, but current technological methods make the integration of the two difficult and time-consuming with significant information loss. The duet multiomics solution +modC kit from biomodal achieves simultaneous genetic and DNA methylation sequencing data at single base resolution from the same DNA fragment, all in a single pre- and post-sequencing integrated workflow. This is achieved with minimal input DNA requirements and avoids the use of harsh chemicals that damage precious samples. The duet bioinformatics workflow performs comprehensive multiomic data analysis to report on all 4 canonical bases as well as methylated cytosine modifications, then integrates these two informational layers to give variant-associated methylation (VAM).
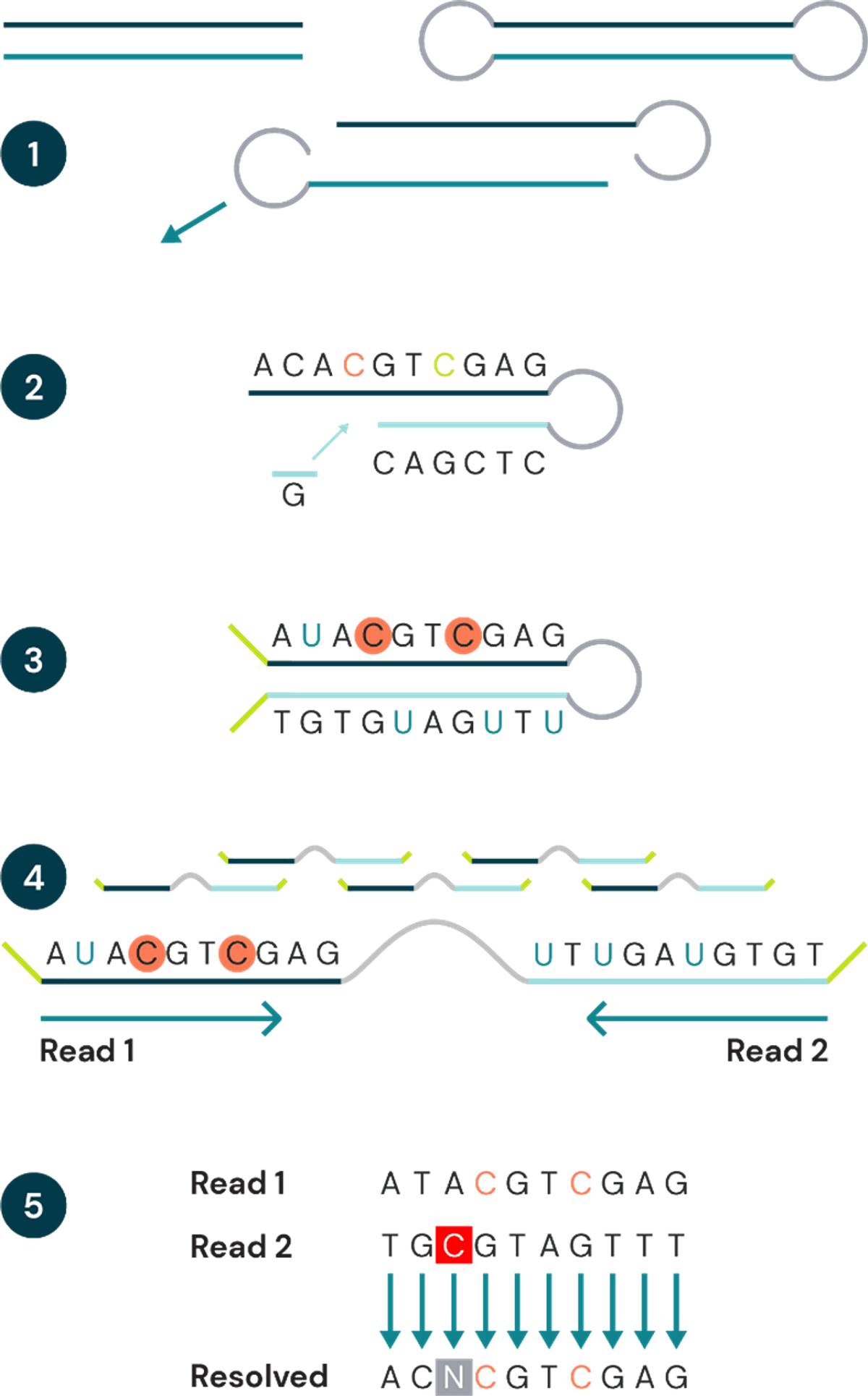
Figure 1 – duet multiomics solution +modC workflow
- Fragmented genomic DNA is ligated to a hairpin adapter at both ends. The hairpin complex is then split into two strands. Both strands move ahead to step 2 (one strand has been omitted for clarity).
- A complementary copy strand is synthesized, accurately capturing the genetic sequence prior to conversion.
- Modified cytosines are protected, and unmodified cytosines are deaminated to uracil. Sequencing adapters are ligated onto strand ends.
- Hairpin complex is linearised and PCR amplified, then sequenced. Read 1 sequences the original strand, read 2 sequences the copy strand. Uracil will be read as thymine.
- Post-sequencing, complementary reads are pairwise aligned. Bases from both reads are resolved using bespoke biomodal pipeline resolution rules to give the original base, plus cytosine modifications, for each position. Sequencing or PCR errors will result in impossible base pairings. These are identified, labelled as N, and filtered out.
biomodal duet multiomics solution +modC run available on BSSH demo data page
To view a NovaSeq 6000 sequencing run using libraries generated from biomodal duet multiomics solution +modC, please visit BaseSpace Sequence Hub. Check our previous blog post on our Demo Data section in BaseSpace Sequence Hub for additional details on how to access published run data.
Below are two links to directly import the runs and project folders into your BaseSpace account. These runs can be found under the “Multiomics” and “Methylation” category. Because these are public data sets, these are free and do not count against storage limits.
To generate this sample data set, seven “Genome In A Bottle” samples were prepared using the biomodal duet multiomics solution +modC kit using 80ng DNA input each, and then sequenced in duplicate at 2x151 on NovaSeq 6000 S4. Samples were loaded onto the sequencer at 250 pM, with 8% PhiX Spike-in.
Run link: https://basespace.illumina.com/s/UalWrnQNZxQ5
Project link: https://basespace.illumina.com/s/MD3iU4v2kn9m
Run assessment
You can use the demo data to compare with your own biomodal duet multiomics solution +modC runs. See our first blog post in this series for additional details on how to evaluate your sequencing run quality.
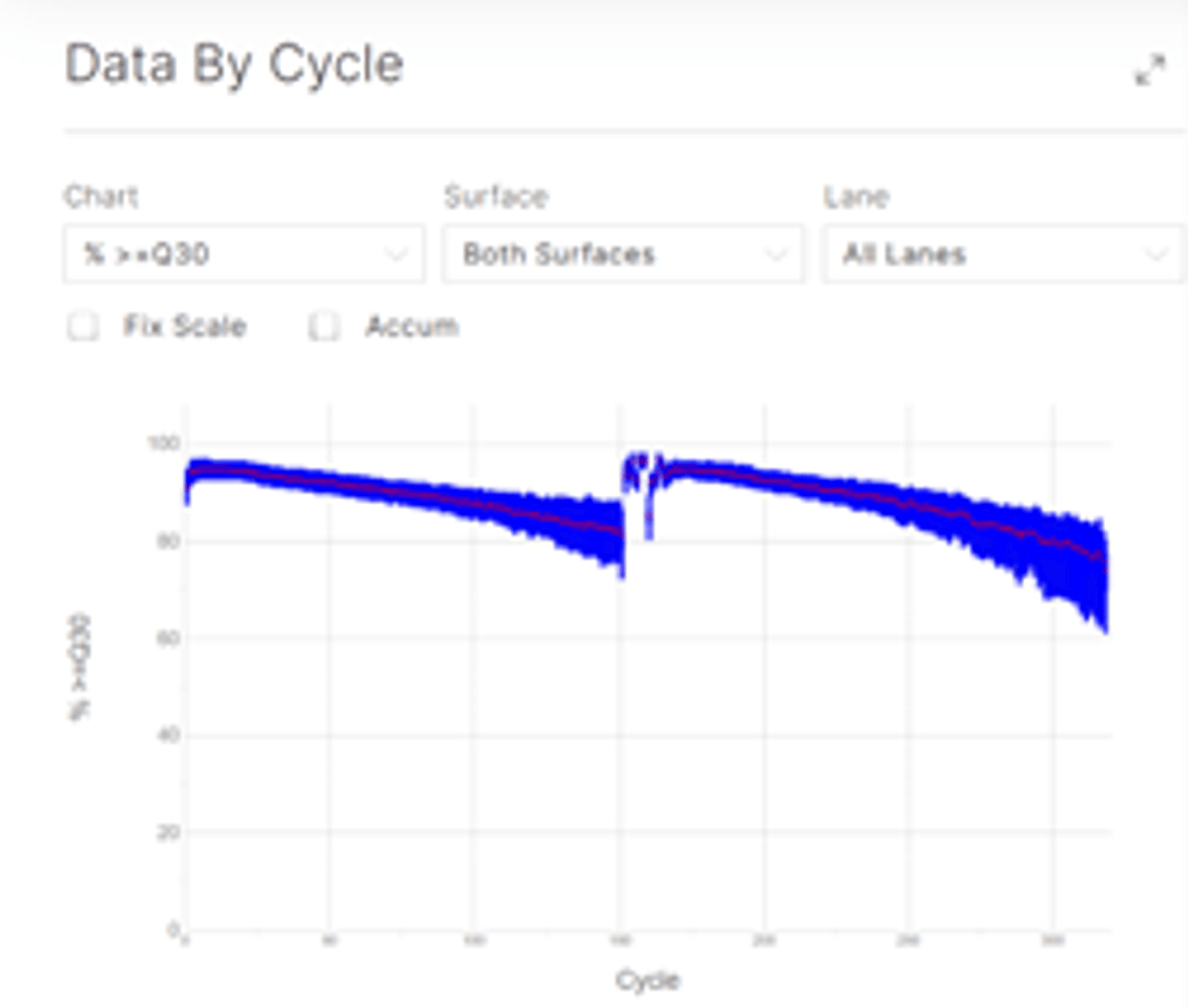
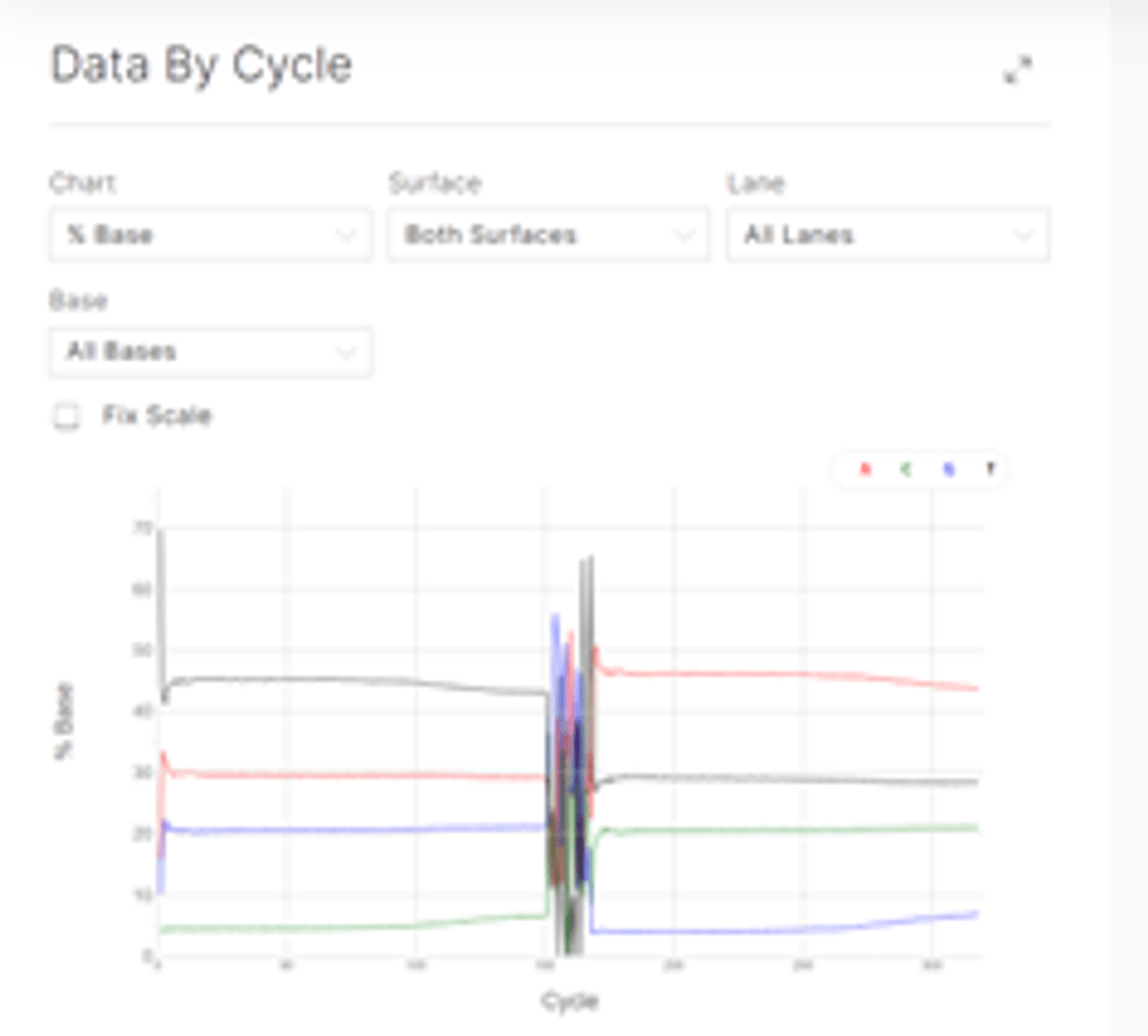
Data analysis
Sequencing data was processed using the biomodal pipeline v1.1.1 and downsampled to ~30X coverage.
The biomodal duet post-sequencing pipeline takes fastQ files directly generated by the sequencer and performs a set of bioinformatic computational steps to produce analysis-ready data in industry-standard file formats. Genetic sequence alignment, methylation quantification, and variant calling are all reported as standard. Additionally, and unique to the duet multiomics solution, Variant-Associated Methylation information is accessible including an Allele-Specific Methylation file.
The following files and reports have been included in the project link for download under “Other dataset” and then “Files”:
- Summary_report – an excel sheet containing summary metrics for all samples analysed. Metrics are associated with reads aligning to the reference genome, and with reads aligning to the spike-in controls. The sheet contains common NGS workflow metrics and duet multiomics solution +modC specific metrics.
- MultiQC – an html report including plots of the most important metrics for all samples including coverage, GC content and methylation bias.
- Allele_specific_methylation – Methylation on reads associated with each allele is quantified and a call of ASM is provided where asymmetry is significant.
- Genome_modC_quantification – files reporting methylated cytosine content for each sample across multiple genomic contexts (CpG, CHG, CHH)
- Variant_call_files – VCF file containing information about variants detected at specific positions in the reference genome.
Example report
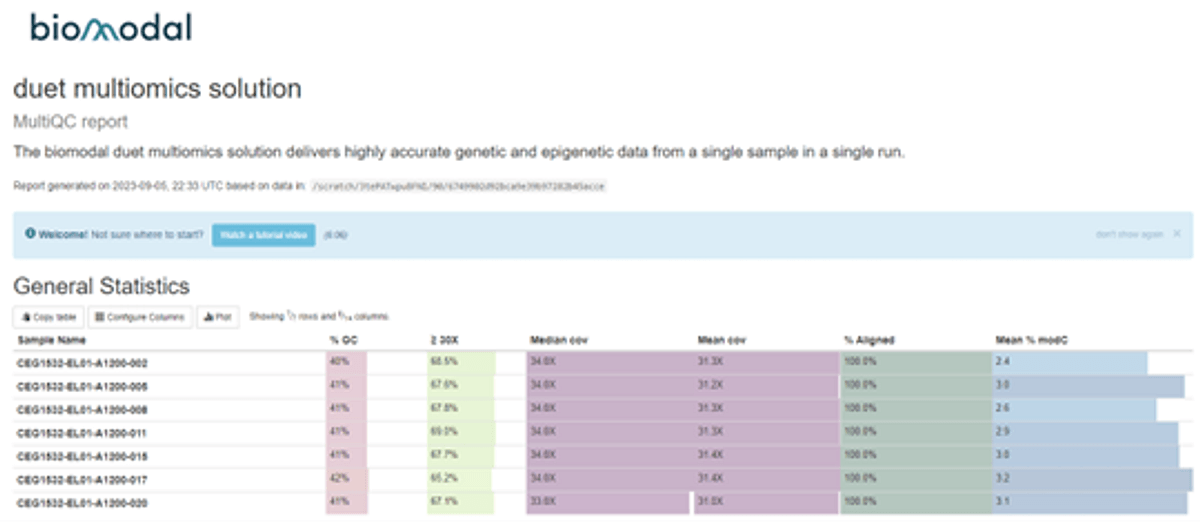
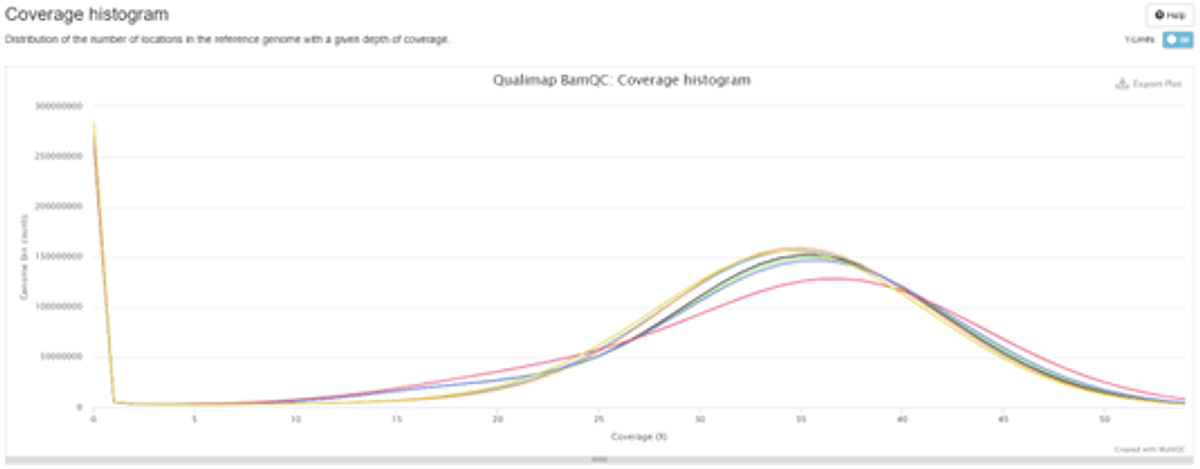
Figure 3 - MultiQC report example
This example screenshot shows some of the information that the QC report contains, including coverage, %GC, and methylation fraction. The interactive html report offers these and a range of other important metrics in simple graphical formats, customisable for your research requirements.
Special thanks to our colleagues at biomodal for providing the data. For further information about library prep and analysis, contact: info@biomodal.com. Contact Illumina Tech support for sequencing related questions.
This material is provided for informational purposes only, to provide an example of the range of capabilities offered by Illumina’s innovative instruments and technologies relating to the analysis of genetic variation and function. It is not intended to be, and should not be construed as, an endorsement of specific third-party products, technologies, or services. Any data presented is neither owned nor verified by Illumina. Customers should independently determine whether using any third-party solutions on Illumina platforms is appropriate for their workflow or laboratory.
For Research Use Only. Not for use in diagnostic procedures. M-GL-02577