- Home
- News & Updates
- DRAGEN v3.5 Updates
-
DRAGEN
-
News
- 03/10/2020
DRAGEN v3.5 Updates
Authored by Angel Pizarro - Associate Director, Product Management
2019 was a big year for DRAGEN. We announced our partnership with the Broad Institute, released several new pipelines, and saw step-function improvements in terms of both accuracy and speed gains across the board.
Looking at our 2020 roadmap, this pace of innovation will continue to accelerate, and we’re excited to share the first 2020 release with you, v3.5.
From a DRAGEN’s-eye view, here’s what’s new in v3.5:
- DRAGEN v3.5 is an integral part of the new NextSeq 1000 and NextSeq 2000 sequencing platform (We felt this one deserved its own post – check it out here)
- Accuracy improvements in somatic, structural variant, and RNA pipelines
- New random and non-random UMI read collapsing capabilities drastically reduces read errors in TSO500 workflows
- New population level cohort analysis capabilities
- support for CNV analysis on Somatic samples
A comprehensive list of new features and details can be found in the DRAGEN v3.5 release notes and recorded webinar.
Today we will focus on just two of these improvements: RNA Fusion detection; and population-level joint calling.
What’s new: RNA Fusion Detection
To address the challenges with RNA-Seq fusion detection, we created a repository of 153 samples with known validated or expected fusions from 5 different sources representative of real-world applications. The sources included libraries from whole RNA and enrichment cell lines, fresh leukemia, and FFPE sarcoma, resulting in 223 expected fusions from 153 samples. Some of these fusions are based on the type of cancer but may be absent from any given sample. There may also be additional fusions that are unverified.
Using the data from TruSeq RNA whole RNA cell lines, we benchmarked our fusion detection against both STAR+Salmon, and DRAGEN v3.4. We found that DRAGEN v3.5 is now 4.5 times faster than STAR+Salmon, and almost twice as fast as the previous version of DRAGEN. We also see improvements in sensitivity, with 38% fewer missed fusions and 20% fewer additional calls from the validated set when compared to DRAGEN v3.4
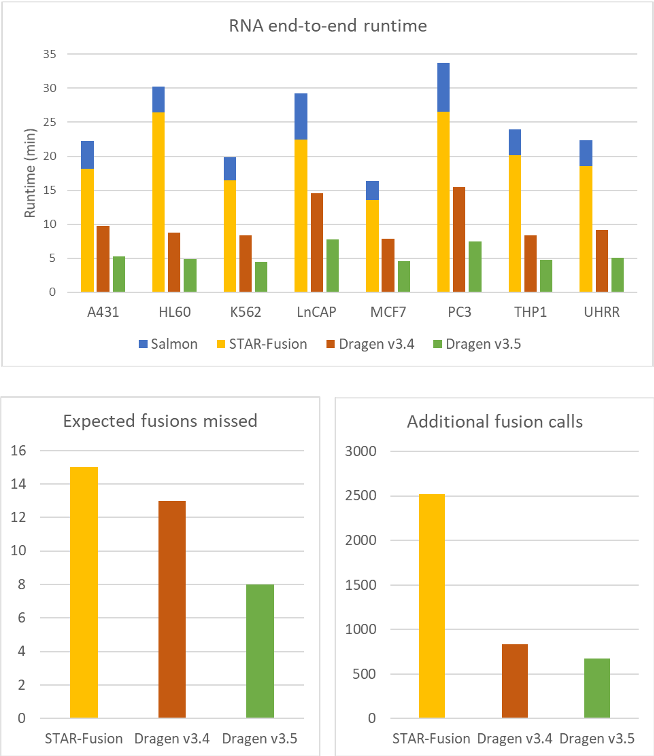
Figure 1. DRAGEN RNA fusion detection is much faster and more accurate than STAR + Salmon
New Population Level Joint Variant Calling
DRAGEN v3.5 has drastically improved its joint calling capabilities beyond a small number of related samples such as trios. DRAGEN is now able to build multi-sample gVCF of up to ten thousand samples within a single server. The output of the DRAGEN gVCF genotyper tool is then be re-analyzed by the DRAGEN joint genotyper to improve calls in low-coverage areas. Maintaining a core value, these algorithms are fast, with the ability to process over 1000 exomes in about 8 hours, and 1000 whole genomes in about 24 hours, all on a single server.
For population level projects that today reach into the hundreds of thousands of individuals (and one day millions) we also developed a CPU-only joint caller that can automatically span multiple compute nodes to distribute the work necessary for these extremely large cohorts.
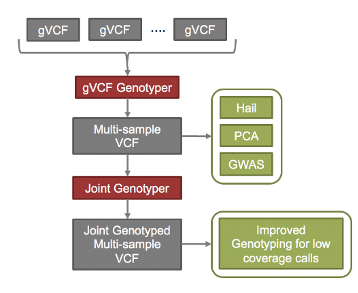
Figure 2. The DRAGEN gVCF genotyper creates a multi-sample gVCF for GWAS tools such as Hail. the DRAGEN JointGenotyper takes this output to produce population-based genotype refinement for improved calls in low-coverage regions.
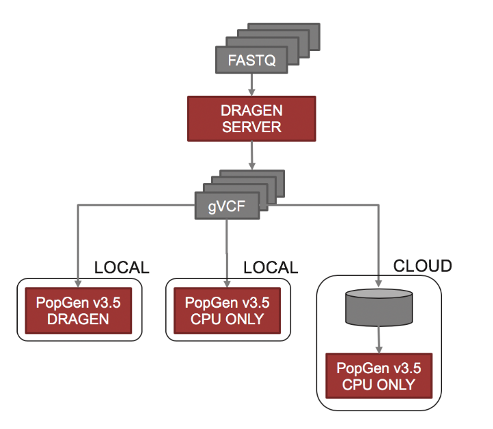
Figure 3. The DRAGEN population level joint genotyper is able to span multiple local or cloud resources in order to analyze extremely large cohorts.
Summary
While officially it’s the Year of the Rat, for us 2020 is the Year of the DRAGEN – and we’re excited to keep delivering new features and pipelines to our customers. As always, we’d love to hear your feedback, and I welcome you to reach out directly. You can get the full release notes for DRAGEN version 3.5 and the software on the DRAGEN software release page at DRAGEN support site. You can also view a pre-recorded webinar on all of the new features in version 3.5 here.
For research use only.