- Home
- News & Updates
- Introducing DRAGEN Original Read Archive (ORA)
-
DRAGEN
-
News
- 03/17/2021
Introducing DRAGEN Original Read Archive (ORA)
Learn more about the DRAGEN v3.8 release, including DRAGEN ORA by attending the webinar on March 23, 2021.
There’s been an estimated 50-fold growth of data in biotech between 2010-20201; in 2019 alone, the global install base of Illumina sequencers generated 150 Petabases of data, which is the equivalent of 500 years of continuous video recordings in high definition. That’s a lot of Netflix! A single 30x human genome has a 50 to 70 GB storage footprint (stored under fastq.gz). Storage costs can quickly stack up as more and more samples are sequenced, and clinical data retention policies require extended storage periods. For example, storing 20,000 whole human genomes for 3 months of hot storage, and 3 years of cold storage in Amazon Web Services (AWS) would cost ~$200,000 (assuming 50 GB per human genome).
In July of last year, Illumina acquired Enancio, an early-stage software company boasting a novel lossless compression technology of genomic data that is a unique combination of fast run times, high compression ratios, and low memory requirements. The wide availability of this technology will have enormous benefits for anyone managing genomics data. Remember that $200,000 storage bill for 20,000 whole human genomes? When stored under the compressed file format, the FASTQ storage bill could go down to as low as $40,000. That’s an 80% direct savings on storage costs and energy consumption.
Enancio’s compression technology, now called DRAGEN ORA (Original Read Archive) is being integrated across the Illumina software portfolio. Four months after the announcement of the acquisition, DRAGEN ORA compression was made available onboard our latest sequencer, the NextSeqTM 1000/2000. And now, with the release of DRAGEN v3.8, this technology is available for on-premise and AWS DRAGEN customers.
Why are we so excited to introduce DRAGEN ORA?
- It’s lossless. No information contained in the FASTQ file is lost, not even the read order. The MD5 checksums of the raw FASTQ file before compression and after compression/decompression are identical.
- It’s fast. It takes ~8 minutes to compress a 30x human genome. If you’re not using DRAGEN and need to decompress a file, that takes ~3 minutes with the standalone decompression SW.2
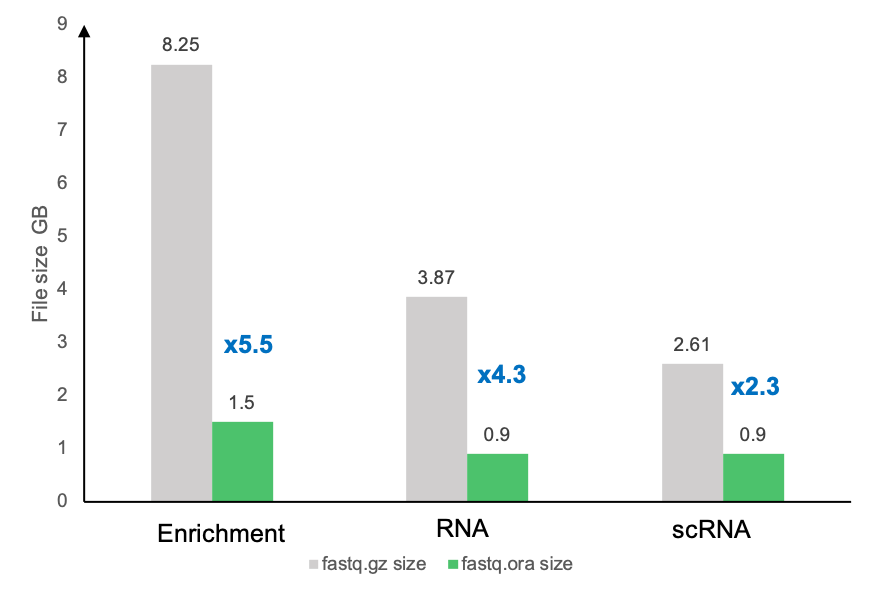
- It achieves high compression ratios. FASTQ file sizes are reduced up to 5x (Figure 1) when stored as fastq.ora (compressed with DRAGEN ORA) compared to fastq.gz. Note that the compression ratios achieved vary by input data type (application, sequencing platform used, species of data). The best compression ratios are achieved with human data on Illumina’s latest sequencers – NextSeq 1000/2000 and NovaSeqTM 6000.
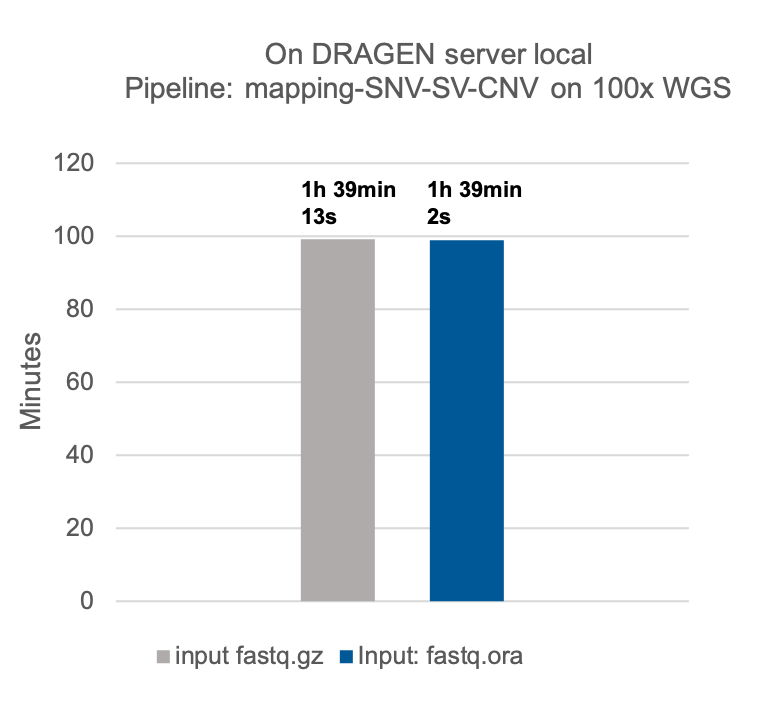
- It’s integrated into DRAGEN. The DRAGEN mapper can natively ingest compressed fastq.ora files, so a separate decompression step is not needed when working with compressed files. Minimal to no run time is added to downstream DRAGEN pipelines when using a fastq.ora file as input.
- It’s included in the cost of your NextSeq 1000/2000, AWS DRAGEN license, or on-premise server license.
- Decompression is freely available. Decompression is integrated within DRAGEN, but what if you want to share a fastq.ora file with a collaborator who doesn’t have access to DRAGEN? We’ve made the decompression software freely available (no license required), so anyone can read a fastq.ora file. The output of the decompression software can be piped to other mappers such as BWA3, Bowtie4, and STAR5 with a single command.
- It’s here to stay. Illumina is committed to ensuring the longevity of the fastq.ora file format so customers can adopt this technology without the fear of not being able to use their compressed files in the future. We will open the file format, allowing anyone to implement their own decompressor.
To learn more about how you can save with DRAGEN ORA, contact your Illumina team.
To learn more about the DRAGEN v3.8 release including DRAGEN ORA, watch the webinar here >>
- https://insidebigdata.com/2017...
- Internal data on file, 2021
- Li, Heng. "Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM." arXiv preprint arXiv:1303.3997 (2013).
- Langmead, Ben, and Steven L. Salzberg. "Fast gapped-read alignment with Bowtie 2." Nature methods 9.4 (2012): 357.
- Dobin, Alexander, et al. "STAR: ultrafast universal RNA-seq aligner." Bioinformatics 29.1 (2013): 15-21.