- Home
- News & Updates
- DRAGEN™ v4.0- Unlock The Full Potential of Genomics
-
DRAGEN
-
Product updates
-
News
- 07/27/2022
DRAGEN™ v4.0- Unlock The Full Potential of Genomics
-
Allison MillerSoftware Product Marketing Manager

DRAGEN v4.0 Software is here! Unlock the power of genomics with comprehensive secondary analysis, from cell biology to pharmacogenomics, all in one single platform. DRAGEN 4.0’s expanded comprehensiveness paired with accuracy and efficiency improvements pave the way for Whole-Genome Sequencing (WGS) secondary analysis at scale. Access the latest software and learn more about new features in our release notes.
Innovation is at the core of what we do at Illumina. DRAGEN 4.0 is our most comprehensive secondary analysis solution yet, with new features enabling our customers to expand their research into key areas such as oncology, pharmacogenomics (PGx), single-cell sequencing, and population genomics all in a single platform. In comparison, it would take over 30 open-source tools to partially replicate the breadth of functionality within DRAGEN 4.01.
Additional accuracy and efficiency improvements make DRAGEN more accurate, comprehensiveness, and efficient than ever before. Let’s take a deeper dive into a few of the new features that make DRAGEN 4.0 so exciting:
Machine Learning Enabled by Default
DRAGEN v3.10 added a powerful and accuracy-standard setting machine learning pipeline as an option within the germline small variant workflow. DRAGEN 4.0 now enables machine learning by default, giving users access to the same small variant calling accuracy improvements out of the box.
The Machine Learning (ML) model is now packaged as part of DRAGEN and supports human samples (hg19/hs37d5 and hg38) for WGS or WES workflows.
Accuracy improvements include:
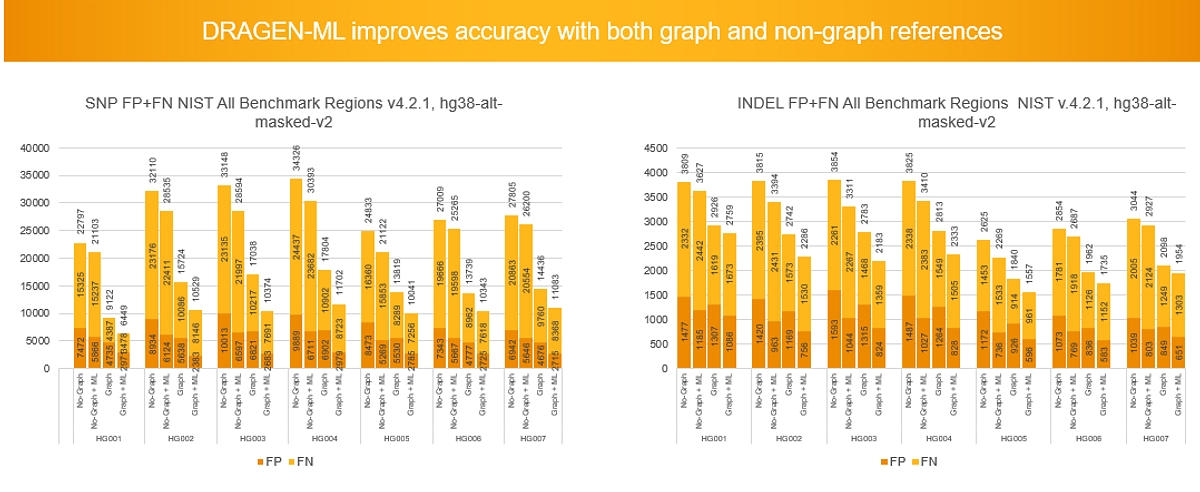
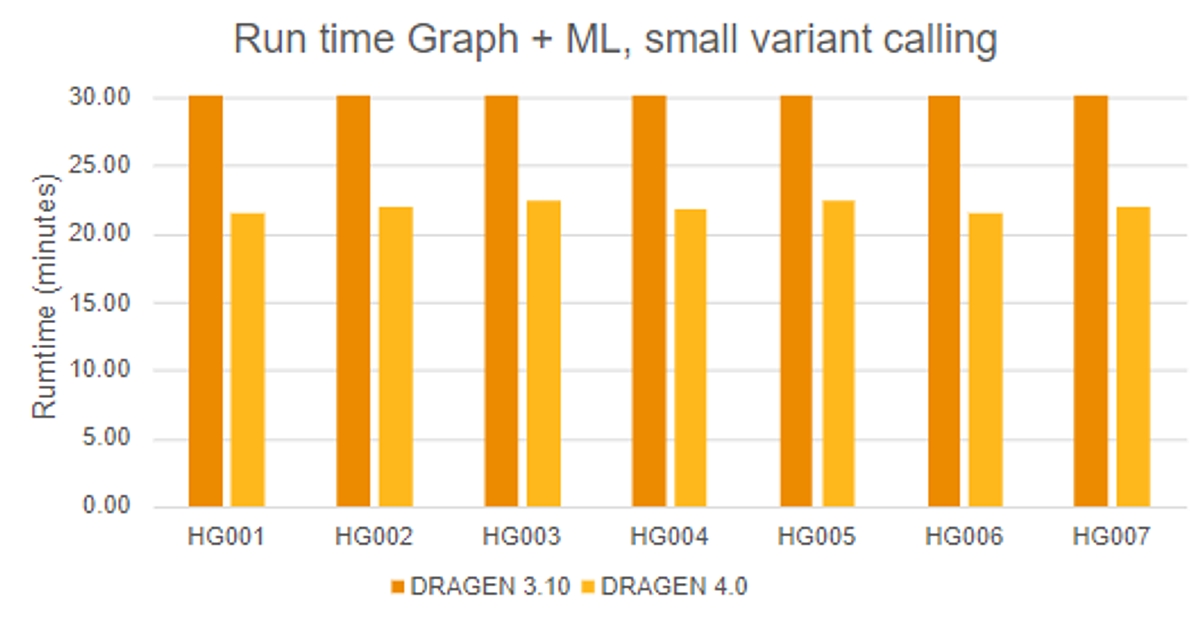
DRAGEN 4.0 Machine Learning also demonstrates increased efficiency, with run times improving by ~26% for small germline variant calling compared to DRAGEN 3.10 ML. Improvements by reference include:
New PGx Star Allele Calling
DRAGEN v4.0 brings significant PGx research capabilities, enabling the calling of 20 Tier 1* genes (CPIC Level A genes). Expansion of DRAGEN PGx research capabilities further supports the genotyping of complex PGx genes and powers insights for optimal genotype and corresponding metabolism status.
New PGx tools include:
- New DRAGEN star allele caller, which calls star alleles and metabolism status for 16 genes
- Targeted callers for CYP2D6 and CYP2B6, which call star alleles and metabolism status for these difficult genes
- New targeted caller for HLA, which outputs the genotype
DRAGEN star allele caller identifies the genotypes of the following 16 Tier-1 PGx genes: CACNA1S, CFTR, CYP2C19, CYP2C9, CYP3A5, CYP4F2, IFNL3, RYR1, NUDT15, SLCO1B1, TPMT, UGT1A1, VKORC1, DPYD, G6PD, MT-RNR1. For each gene, the caller returns the optimal genotype along with the corresponding metabolism status that is associated with that genotype.
DRAGEN 4.0 now additionally calls for CYP2B6 as a part of the star allele caller, an important but challenging gene complicated by sequence homology with pseudogene paralog CYP2B7. A new HLA genotyping algorithm is introduced as well, inferring HLA genotypes.

*Tier 1 = CPIC Level-A genes as of March 1, 2022
Expansion Hunter v5
Short Tandem Repeat (STR) expansions are a major cause of over 20 severe neurological disorders. DRAGEN includes a repeat-expansion detection method, known as Expansion Hunter, to maximize WGS and power genetic disease research insights.
DRAGEN 3.10 brought Expansion Hunter v4, supporting genotyping of 30 STR loci. DRAGEN 4.0 now integrates Expansion Hunter v5, which expands the default catalog 2x and supports ~50k additional loci, including BEAN1, a challenging gene implicated in Spinocerebellar Ataxia.
Expansion Hunter v5:
- Expands the default catalog supported to 60 pathogenic STR loci (including 30 from gnomAD), including genes such as DMD, implicated in muscular dystrophy.
- Enables users to optionally call repeats on an expanded catalog with ~50K polymorphic STR loci in DRAGEN 4.0 with minor runtime impact
- Minimizes runtime impact and enables scaling to large STR catalogs. With other variant callers, EH adds negligible overhead with default catalog and ~8-10 min overhead with expanded catalog on f1.4xlarge
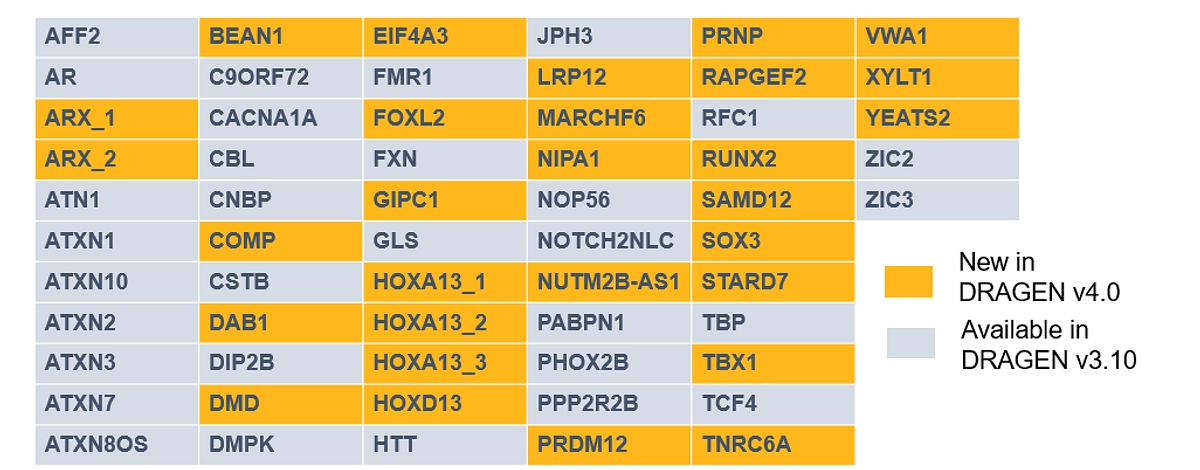
New End-to-End Imputation Pipeline
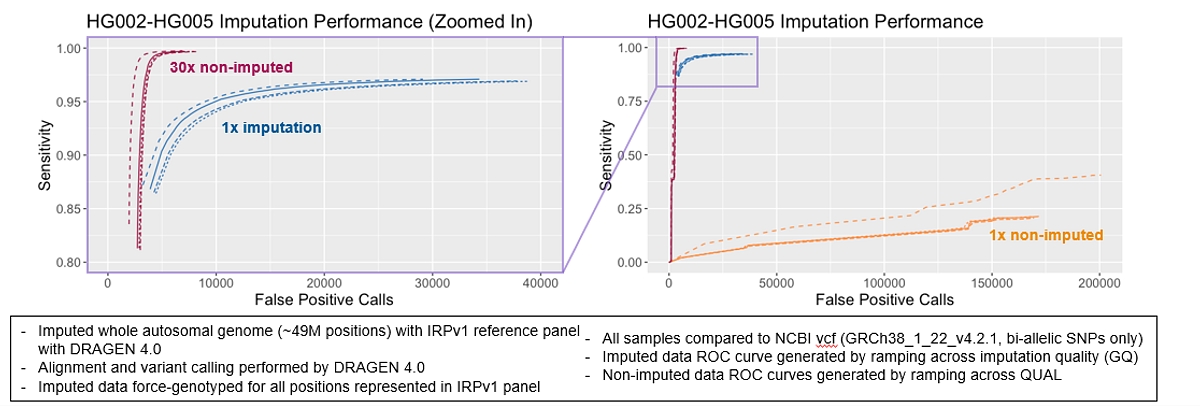
To truly unlock the full potential of genomics, it’s crucial that we make WGS more accessible to a broader audience. DRAGEN 4.0 improves WGS accessibility by introducing a new end-to-end Imputation Pipeline, enabling accurate genotyping of human reference, low-pass sequencing data.
DRAGEN 4.0 integrates the AVX accelerated GLIMPSE1 imputation tool and is 1.7x faster than original GLIMPSE code. The new DRAGEN Imputation pipeline is:
- An end-to-end pipeline: a user-friendly tool to execute scalable whole genome imputation
- Able to impute a single sample or supply a list of VCF files (.txt file) to impute <=100 samples simultaneously
- BaseSpace™ Sequence Hub available via DRAGEN Imputation app
Using the DRAGEN Imputation end-to-end pipeline, analytical sensitivity of 1x data improves by >=70% and reduces false positives by >=80%.
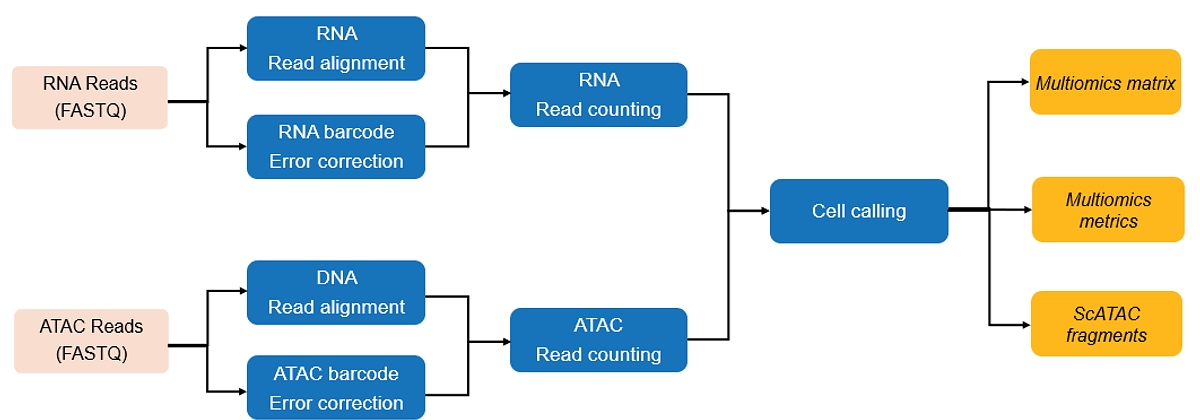
New Multiomics Single-Cell Pipeline
DRAGEN 4.0 features also expand analysis capabilities for our cell and molecular biology customers, specifically in single-cell experiments. DRAGEN 4.0 introduces two new exciting pipelines: single-cell ATAC-Seq and single-cell multiomics.
The DRAGEN single-cell ATAC-Seq pipeline enables single-cell resolution profiling of chromatin accessibility, and now can be run in combination with DRAGEN’s single-cell RNA-Seq pipeline as part of the single-cell multiomics pipeline. DRAGEN’s new single-cell multiomics pipeline allows the processing of combined single-cell ATAC and single-cell RNA datasets generated from the same set of biological cells to enable simultaneous profiling of the transcriptome and epigenome.
The DRAGEN single-cell multiomics workflow at a glance:
Other Key Features
Additional DRAGEN 4.0 features include:
- Structural Variant (SV) Calling Runtime and Accuracy Improvements: DRAGEN 4.0 improves insertion recall by 2% compared to DRAGEN v3.10, and brings additional runtime improvements, with optimizations leading to improved analysis times across multiple SV workflows.
- Build Your Own Custom Multigenome Reference: DRAGEN v4.0 now enables you to build your own alt-aware population hash table “graph” reference genome.
- BCL Convert Updates – Feature parity with bcl2fastq. BCL conversion to FASTQ.ORA is now more user friendly and can be enabled in a single command.
- HLA typing accuracy improvements: A new HLA typing algorithm is introduced, using nucleotide-based-HLA specific alignments and improved probabilistic modeling to infer HLA genotypes.
- Fragmentomics: The new DRAGEN Fragmentomics pipeline computes three metrics to quantify genome-wide fragment profiles for ctDNA assays with high efficiency.
- High coverage somatic analysis: Improved efficiency enabling support of WGS samples with an coverage of up to 1000x
- Tumor only SV improvements: Accuracy improvements with scoring model and systematic noise filtering
- Methylation Improvements: Additional improvements to the DRAGEN methylation pipeline include an end-to-end dedup as well as UMI support
Learn More
The DRAGEN 4.0 release is packed full of comprehensiveness, accuracy, and efficiency gains, and this blog has only touched the surface. For an in-depth summary of all DRAGEN 4.0 features, view our user guide.
Visit the DRAGEN page to learn more about our technology and other updates, including our latest v4 server release.
Other Resources:
1. Illumina Data on File, 2022
For Research Use Only. Not for use in diagnostic procedures
- 2