- Home
- News & Updates
- DRAGEN v4.5: Illuminating the Dark Genome with Advanced Variant Detection
DRAGEN v4.5: Illuminating the Dark Genome with Advanced Variant Detection

Within the genomics field, it is widely acknowledged that certain genomic regions remain challenging to analyze using short-read sequencing technologies. Consequently, analyses reliant on short reads encounter several recognized limitations: formalin-fixed, paraffin-embedded (FFPE) tumor samples often introduce significant technical variability; haplotype phasing generally necessitates either family trios or costly long-read sequencing; and structurally complex or segmentally duplicated regions are difficult to resolve without the use of specialized assays.
DRAGEN v4.5 rewrites these rules.
This DRAGEN release does not just incrementally improve the secondary analysis platform’s industry-leading accuracy, it fundamentally expands what researchers can reliably extract from standard WGS data. By combining breakthrough Illumina TruPath™ Genome technology, advanced machine learning (ML) across germline and somatic workflows, and comprehensive oncology research enhancements, DRAGEN v4.5 enables discovery in regions and samples that were previously beyond reach.
With these advances, researchers can now more confidently analyze medically relevant genes in segmental duplications, detect low-frequency somatic variants in noisy FFPE samples, phase haplotypes spanning millions of bases, and identify oncovirus integration sites, all from the same simple short-read WGS workflow and analyzed in hours instead of days.
Let us explore how DRAGEN v4.5 achieves this transformation and why it matters for users upgrading from earlier releases.
Germline Workflow Improvements: Unmatched Accuracy Through Personalization
DRAGEN v4.5 significantly reduces the noise in germline calling, delivering a 20% reduction in small variant errors compared to v4.4.* It expands on the pangenome and provides significant genotyping improvements on complex loci, for medically relevant genes like CYP21A2, GBA, SMA silent carrier and 4-filed HLA typing.
Expanded Pangenome
DRAGEN v4.5 continues to make the pangenome reference more inclusive by adding more ancestries. This release adds Middle Eastern ancestry to the pangenome reference for the first time through the addition of 16 representative samples, expanding the reference to 144 globally diverse samples across 27 ancestries and improving population representation and mapping accuracy across diverse genomes.
Personalized reference by default yields unmatched germline small variant accuracy
Compared to DRAGEN v4.4, DRAGEN v4.5 delivers a substantial improvement in germline small variant accuracy, reducing total SNP and indel false positives and false negatives by ~20% on HG002 when benchmarked against the NIST T2TQ100 truth set (See Figure 1). These gains are driven by personalized pangenome‑based mapping and enhanced germline variant calling, with consistent error reduction observed across both SNPs and indels. This results in higher‑confidence germline call sets, fewer spurious variants requiring manual review, and improved performance in difficult‑to‑map genomic regions,while maintaining comparable runtime for 34× WGS analyses (≈35 minutes across callers).
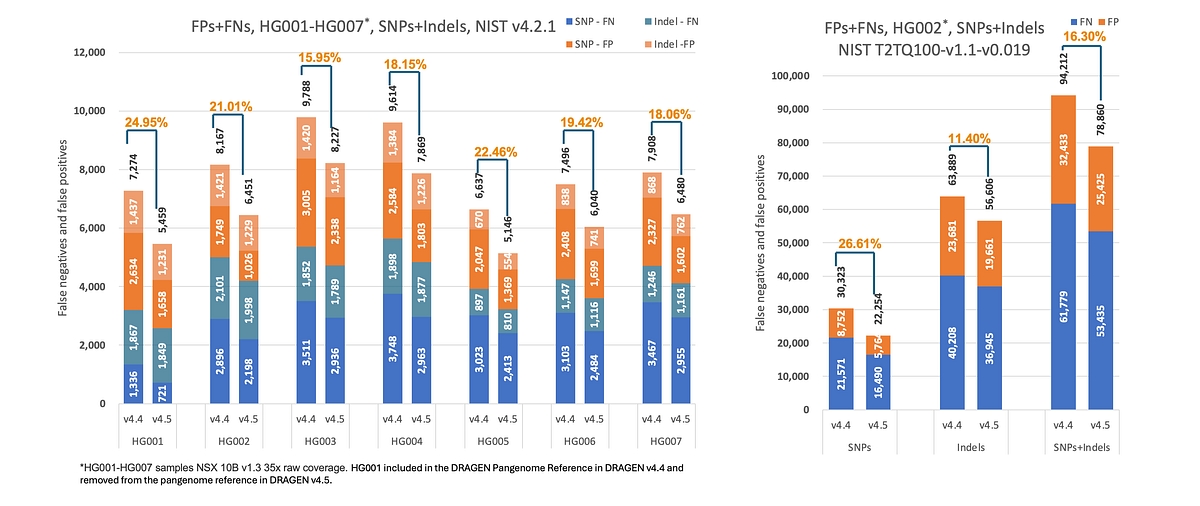
Figure 1: 20% reduction in germline SNP and indel errors with DRAGEN v4.5 compared to v4.4 on HG002, benchmarked against the NIST T2T‑Q100 truth set in high-confidence regions.*
SMN Caller Updates
The new ML‑driven SMN1 duplication haplotype caller in DRAGEN v4.5 substantially improves detection of SMA 2+0 silent carriers, a class of carriers missed by copy‑number–only methods. Compared to the legacy single‑SNP approach (NM_000344.3:c.*3+80T>G), DRAGEN’s biomarker‑based caller achieves 26% higher recall and 33% greater precision in a 1K Genomes–derived evaluation (See Table 1). This improvement is particularly impactful for populations with higher silent carrier prevalence, such as individuals of African ancestry, where single‑marker screening has limited analytical sensitivity. These gains translate to more reliable carrier detection, reduced false positives, and more equitable performance across ancestries using standar WGS or WES data.
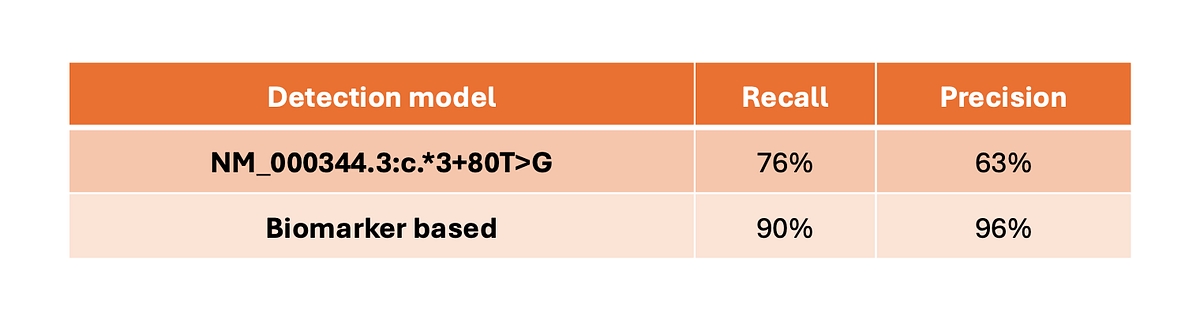
Table 1: Performance comparison of SMN1 silent carrier detection, tested over 3202 1kg samples, using the legacy single‑SNP marker (NM_000344.3:c.*3+80T>G) and the DRAGEN v4.5 biomarker‑based SMN1 duplication haplotype caller.*
Other notable improvements in the Germline workflow:
- Further improved SV accuracy with 5% f-score gain
- Improved mitochondrial SV calling, with accurate heteroplasmy estimation
- CNV caller low pass WGS mode 1-10x coverage
- Improved CYP21A2 genotyping accuracy
- HLA typing with 4-field resolution
Oncology Research Workflow Enhancements
Somatic workflows in DRAGEN v4.5 are designed to handle the most challenging real-world samples, such as FFPE and liquid biopsy, without increasing computational burden. This release introduces ML-driven somatic variant calling, leading to significant reduction in FFPE artifacts, comprehensive oncovirus detection and mutational signatures reporting.
ML‑driven somatic variant calling (as a non-default option)
DRAGEN v4.5 introduces ML‑driven somatic small‑variant calling, improving analytical sensitivity and specificity across a wide range of tumor types. By learning complex signal patterns in noisy data, this approach improves true variant detection while maintaining high precision even in challenging sample types without increasing runtime. As shown in Figure 2, ML‑driven somatic SNV calling in DRAGEN v4.5 consistently reduces total errors relative to the non‑ML v4.5 caller and delivers the highest analytical sensitivity across all four benchmark cell lines, including the latest NIST HG008 benchmark [1]. In contrast to third‑party tools, DRAGEN exhibits markedly fewer false‑negative errors, driving superior sensitivity without inflating overall error counts.
Control your workflow: to ensure a seamless transition for existing production pipelines, Somatic ML is disabled by default in this release. Users can easily enable these advanced detection models using a simple command line option to unlock the highest level of analytical sensitivity and artifact suppression.
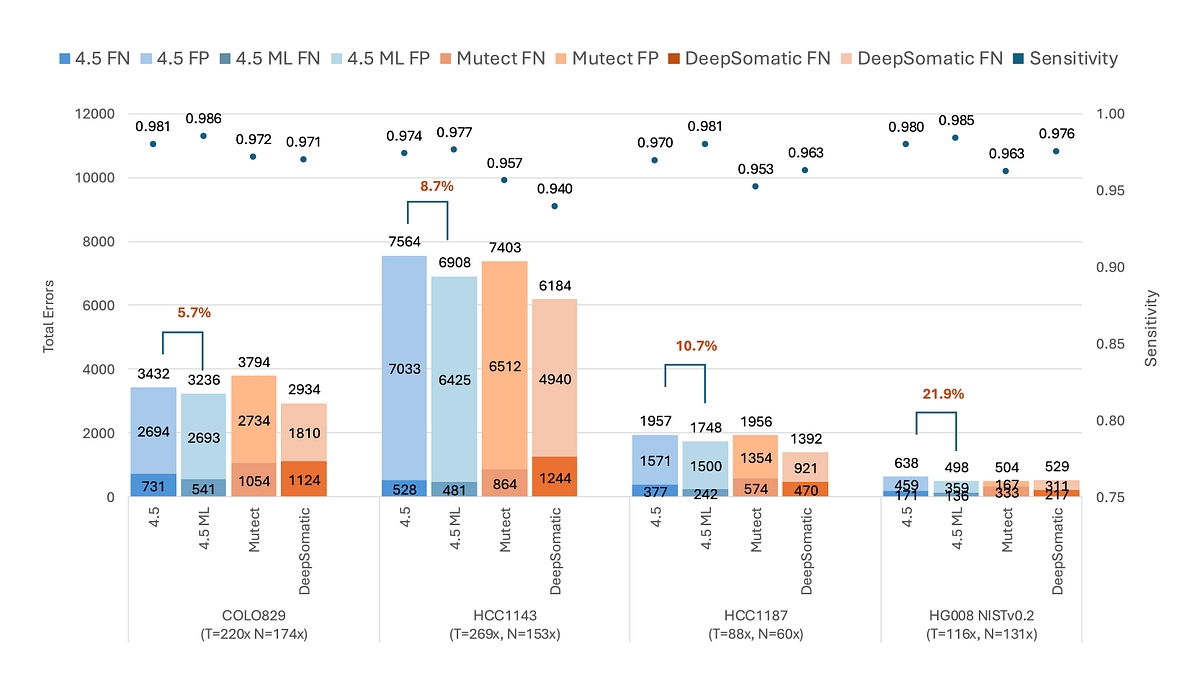
Figure 2: Somatic SNV calling performance across four benchmark cell lines comparing the ML‑driven DRAGEN v4.5 caller, the non‑ML DRAGEN v4.5 caller, and third‑party variant callers. [2]
FFPE noise reduction
Somatic variant calling in DRAGEN v4.5 includes targeted improvements for FFPE samples, reducing artifacts introduced during sample preparation. Using optional ML‑based somatic score recalibration, DRAGEN v4.5 dramatically suppresses FFPE‑driven false positives without relying on rigid hard filters. In a normal‑vs‑normal FFPE benchmark where all calls are artifacts, ML reduces false positives by 93.4% for SNVs and 87.4% for indels (See Figure 3). This substantial reduction effectively eliminates the FFPE noise floor, enabling cleaner VCFs and more confident detection of true low‑frequency somatic variants in degraded tumor samples.
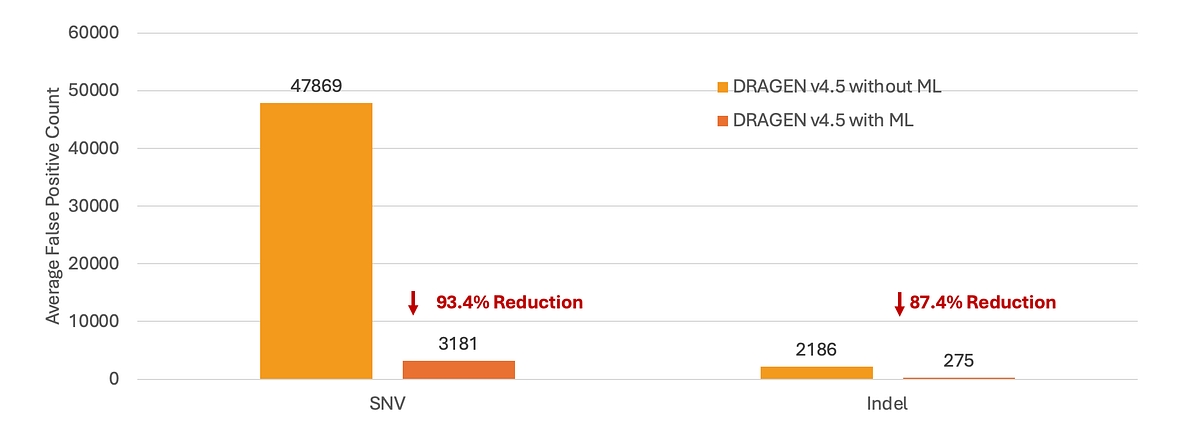
Figure 3: Reduction of FFPE‑induced somatic variant artifacts using ML‑based somatic score recalibration in DRAGEN v4.5.*
Comprehensive oncovirus detection and integration site calling
DRAGEN v4.5 expands support for oncovirus detection, including identification of viral integration sites within the host genome. These capabilities enable more comprehensive characterization of virus‑associated cancers by linking viral presence to structural genomic disruption. In an internal Illumina analysis of 198 tumor samples spanning multiple primary tumor sites, DRAGEN detected 100% of oncoviruses identified by an orthogonal assay, while additionally identifying 18 more oncoviruses due to broader HPV typing support and increased analytical sensitivity for EBV detection (See Table 2). Meanwhile, quantitative viral read counts show strong agreement with independent methods (Pearson r = 0.92), demonstrating trustworthy viral burden estimation.
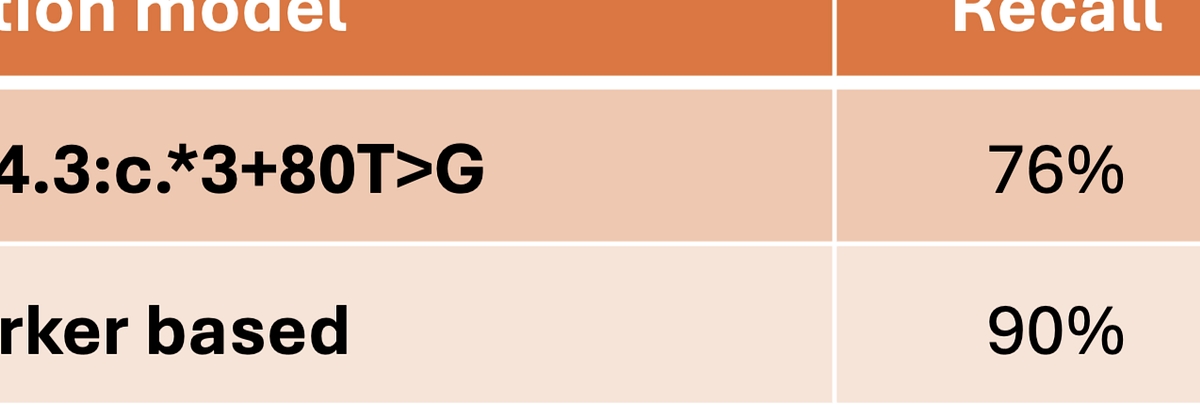
Table 2: Oncovirus detection performance of DRAGEN v4.5 across 198 tumor samples representing multiple primary tumor sites, compared against an orthogonal assay and independent informatics.*
Other notable improvements in the Somatic workflow:
- ML-based somatic small variant calling notably improves recall of low VAF variants (5-10%) outperforming third party tools
- Introducing a Somatic T/N “Fingerprint” mode for WGS and WES used for MRD monitoring in plasma
- Introducing cytogenetic mode in somatic WGS workflow
- Mutational signatures reporting: SBS, DBS, ID
- Bulk RNA: improved analytical sensitivity for WTS, output selected intragenic and read-through fusions by default, PTD detection
TruPath™ Genome – Long-Range Genomic Insight
Another transformative feature of DRAGEN v4.5 is introducing support for the TruPath Genome†, which extends short-read sequencing into long-range genomic insight. By incorporating long-range proximity information directly into DRAGEN’s analysis pipelines, users can now resolve haplotypes, repetitive regions, and structurally complex loci at scale without the cost or workflow complexity of long-read sequencing.
Phasing
TruPath Genome phasing in DRAGEN v4.5 delivers near–long‑read performance from short‑read sequencing, phasing up to 98% of genes with high‑molecular‑weight (HMW) DNA input and 89% with standard DNA input. TruPath generates phase blocks spanning millions of bases (NG50 up to ~6.1 Mb), substantially exceeding PacBio phase block lengths while maintaining high phasing accuracy across both standard and HMW DNA inputs. Despite these long phase blocks, DRAGEN achieves a low Hamming error rate for standard and HMW DNA inputs at 4.08% and 5.26%, respectively, outperforming PacBio’s performance of a 5.90% Hamming error rate in both input types (See Figure 4). This performance enables accurate haplotype resolution and gene‑level variant interpretation at scale from short‑read sequencing, without the cost, throughput constraints, or workflow complexity of long‑read technologies.
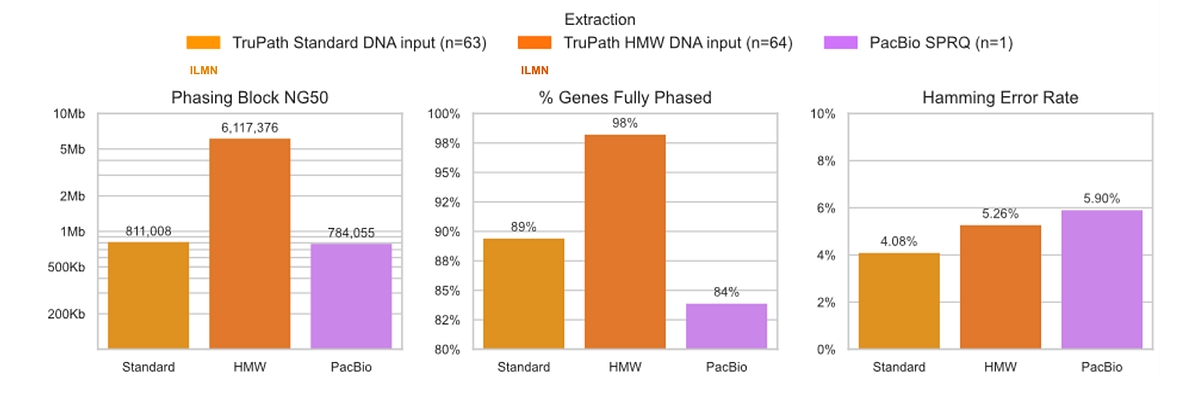
Figure 4: Comparison of phasing performance for TruPath Genome in DRAGEN v4.5 and PacBio across standard and high‑molecular‑weight (HMW) DNA inputs. [3]
Allele-resolved STRs
DRAGEN v4.5 improves STR analysis with TruPath Genome by enabling more accurate length estimation of large and expanded alleles, which are strongly correlated with disease and are often misclassified by conventional short‑read methods. Across loci such as FMR1, DMPK, FXN, and C9ORF72, DRAGEN with TruPath more reliably separates normal, intermediate, premutation, and pathogenic expansion ranges and reports two phased STR alleles per locus, rather than a single aggregated value as in the standard DRAGEN STR workflow (See Figure 5).This allele‑resolved STR representation improves repeat expansion classification from WGS data by preserving allele‑specific repeat lengths.
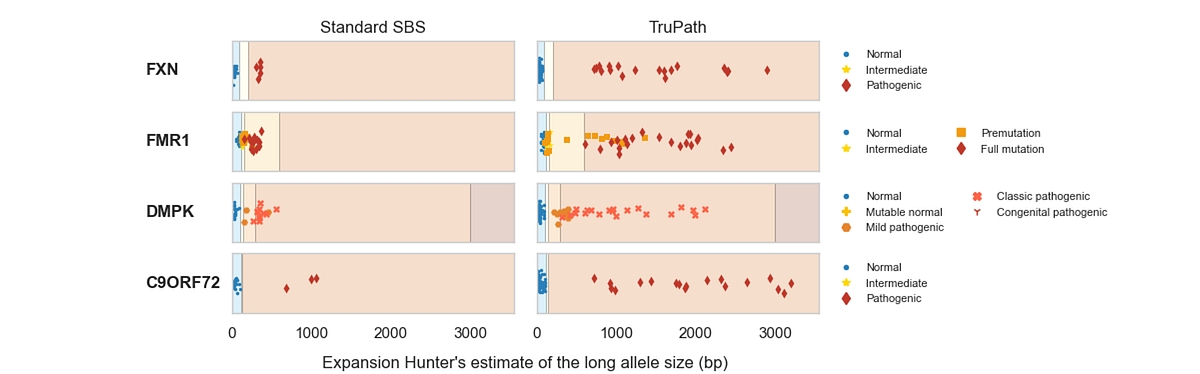
Figure 5: STR length estimates for FMR1, DMPK, FXN, and C9ORF72 comparing the standard DRAGEN STR workflow with TruPath Genome, illustrating allele‑resolved STR reporting across expansion size ranges. [4]
Resolving Segmental Duplication Regions with fully phased haplotypes: multi-region joint detection (MRJD)
DRAGEN v4.5 enhances MRJD by leveraging TruPath Genome read linking long‑range haplotype information to accurately resolve highly homologous and segmentally duplicated regions that confound standard short‑read analysis. MRJD supports a list of 15 medically relevant paralogous genes at launch: PMS2, SMN1/SMN2, NCF1, RCCX (CYP21A2, TNXB), STRC, CYP2D6, CYP11B1/CYP11B2, CFHR1–CFHR4, and USP18.
In the PMS2 locus, which shares ~99% sequence identity with its pseudogene PMS2CL across ~21 kb, conventional short reads collapse signal across paralogs, whereas MRJD in v4.5 cleanly separates gene‑ and pseudogene‑specific haplotypes. As shown in the PMS2 example, DRAGEN reconstructs distinct haplotypes for PMS2 and PMS2CL with concordance comparable to on‑market long‑read technologies, enabling accurate variant attribution without specialized sequencing assays (See Figure 6). This substantially reduces false positives and misassigned variants in biologically important duplicated genes, expanding the set of regions that can be analyzed confidently using standard WGS data.
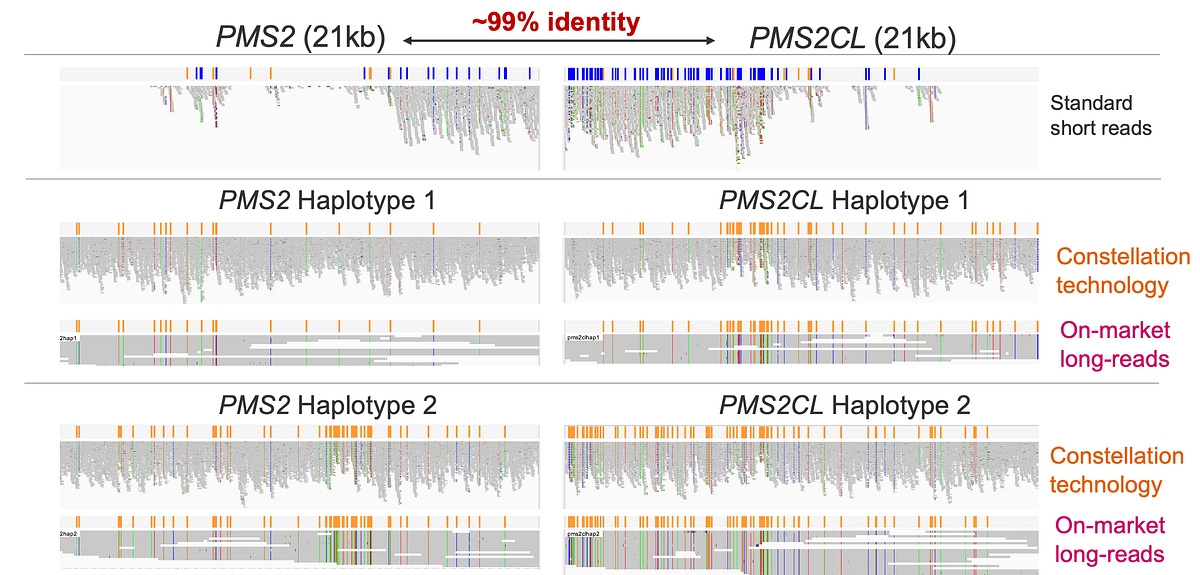
Figure 6: Haplotype reconstruction at the PMS2/PMS2CL locus comparing collapsed short‑read signal with MRJD‑based haplotype separation using TruPath Genome in DRAGEN v4.5 and concordant haplotypes from on‑market long‑read sequencing. Data previously presented at the Advances in Genome Biology and Technology (AGBT) 2026 conference.
Learn More About DRAGEN v4.5
With DRAGEN v4.5, Illumina continues to expand what’s possible with short‑read sequencing, delivering more comprehensive variant discovery, deeper biological insight, and simplified workflows without compromising speed or scale.
To learn more about the new capabilities in DRAGEN v4.5, register for our upcoming technical webinar here, where we will explore these advances in detail and demonstrate how they can be applied across real‑world genomic workflows.
References
[1] MacDaniel J, et al. Development and extensive sequencing of a broadly consented Genome in a Bottle matched tumor‑normal pair. Scientific Data. 2025.
https://www.nature.com/articles/s41597-025-05438-2
[2] Somatic benchmarking reference datasets from COLO829, HCC1143, and HCC1187 two‑technology truth sets from the Lancet2 study, accessed via the following Google Cloud Storage locations:
- gs://lancet2-paper/truths/COLO829/two-tech_truth/COLO829.TwoTechTruth.final.vcf.gz
- gs://lancet2-paper/truths/HCC1187/two-tech_truth/HCC1187.TwoTechTruth.final.vcf.gz
- gs://lancet2-paper/truths/HCC1143/two-tech_truth/HCC1143.TwoTechTruth.final.vcf.gz
The HCC1395 somatic truth set was obtained from the Sequencing Quality Control (SEQC) Consortium via the NCBI Reference Samples repository:
https://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/seqc/Somatic_Mutation_WG/release/latest/
[3] PacBio GIAB trio phasing benchmark dataset (HG002).
https://downloads.pacbcloud.com/public/revio/2024Q4/WGS/GIAB_trio/HG002_rep1/analysis/v3.0.2/
[4] Pathogenic STR expansion thresholds were derived from publicly available STR resources, including STRchive (https://strchive.org) and STRipy (https://stripy.org). All other data shown are Illumina internal data on file.
Footnotes
* Illumina internal data on file
† TruPath Genome refers to Illumina’s short‑read sequencing approach that incorporates long‑range proximity information through specialized library preparation and instrumentation. Support for TruPath Genome analysis in DRAGEN v4.5 requires sequencing on NovaSeq X systems running system software v1.4 or later and use of the TruPath Genome library preparation kit.