- Home
- News & Updates
- Solving the Variant Aggregation Bottleneck to Unlock Genomic Discovery From Cohorts
-
DRAGEN
-
News
- 05/29/2026
Solving the Variant Aggregation Bottleneck to Unlock Genomic Discovery From Cohorts


A GenomeWeb/Illumina Webinar Summary
This GenomeWeb report summarizes a webinar, sponsored by Illumina, in which Catherine Snow, product manager at Genomics England, discussed her team’s experience using the DRAGEN™ Iterative gVCF Genotyper to build a new variant aggregate with double the number of genomic samples and more than 60 million additional detected variants.
Zhuoyi Huang, associate principal bioinformatics scientist, and Ole Schulz-Trieglaff, senior staff bioinformatics scientist at Illumina, joined Snow to provide additional details on DRAGEN and its potential to overcome the limitations of legacy aggregation tools by enabling iterative analysis and batchwise processing without re-genotyping previously sequenced cohorts.
Variant aggregation is a powerful genetic analysis approach that combines multiple DNA variants across numerous genomic samples, increasing the statistical power needed to detect genomic associations with disease. This methodology is particularly beneficial in genome-wide association studies (GWAS) and phenome-wide association studies (PheWAS), where individual rare variants are too infrequent to show significant effects on their own in population-level samples.
SOLVING VARIANT AGGREGATION BOTTLENECKS
Huang kicked off the webinar by discussing bottlenecks in current variant aggregation approaches for large cohorts. Variant aggregation is iterative. New samples are added to existing cohorts, expanding population-level data over time. When this happens, current aggregation software often requires scientists to reprocess or re-genotype previous samples. This is extremely costly, inefficient, and computationally demanding, Huang said. Additionally, many open-source software tools have lower accuracy, with a higher occurrence of false-positive variant calls.
Interoperability is also a challenge for traditional tools. “There are a lot of formats required by different downstream analysis software. Some analyses require a binary format for the genotype, others require gVCF. It’s difficult to have one pipeline that can serve different needs,” said Huang. He also highlighted the need for flexibility across platforms and cloud storage solutions.
Modern variant aggregation tools must be scalable with short turnaround times to accelerate insight discovery, critical for disease research and drug development. Huang introduced listeners to the DRAGEN Iterative gVCF Genotyper, a scalable, high-throughput approach to variant aggregation that includes iterative processing, batchwise efficiency, accuracy at scale, seamless integration, and deployment flexibility.
IMPROVING THE ANALYSIS OF 100,000+ GENOMES
Next, Snow discussed how these features helped power population-scale insights for translational research and eventually for clinical research at Genomics England. Genomics England was established in 2012 to deliver the 100,000 Genomes Project, an ambitious project to sequence 100,000 genomes from patients with cancer and rare diseases in the United Kingdom with the goal of integrating genomic medicine into healthcare. Upon completing the project in 2017, Genomics England created the NHS Genomic Medicine Service, which implements genetic variant detection into routine healthcare using the datasets generated by Genomics England.
“The power of genomics to those researchers who use that data is that we aggregate it together,” said Snow. Aggregates aren’t new to Genomics England. They currently have an aggregate from the 100,000 Genomes Project of patients with rare diseases. “We know from the extensively cited research that it’s our most valuable dataset. However, because it is only from the 100,000 Genomes Project, it comes with its limitations,” said Snow.
She explained that when the project was first set up, Genomics England compared sequenced genomes to an older, now outdated reference sequence for the human genome. As a result, there are older genomes and some newer genomes since the completion of the 100,000 Genomes Project that are not included in their most recent and widely cited variant aggregate. “What that’s resulted in is that our current aggregate is essentially giving us diminishing value and lowering the outcome for participants,” said Snow.
To solve this problem, Snow and colleagues turned to DRAGEN to create a new, more useful variant aggregate. Using DRAGEN, they reanalyzed their legacy germline genomes from the 100,000 Genomes Project as well as more recently acquired germline genomes from the NHS Genomic Medicine Service. This enabled them to standardize all germline genomes in-house as well as across large-scale national and international programs, including the UK Biobank and the US National Institutes of Health’s All of Us Research Program.
“This has been a really successful collaboration with Illumina,” said Snow. Using DRAGEN, her team developed a new variant aggregate with double the number of samples and increased the number of detected variants in their aggregate by 60 million (see Table 1). The improved variant aggregate will be available to the research community to use in 2026.
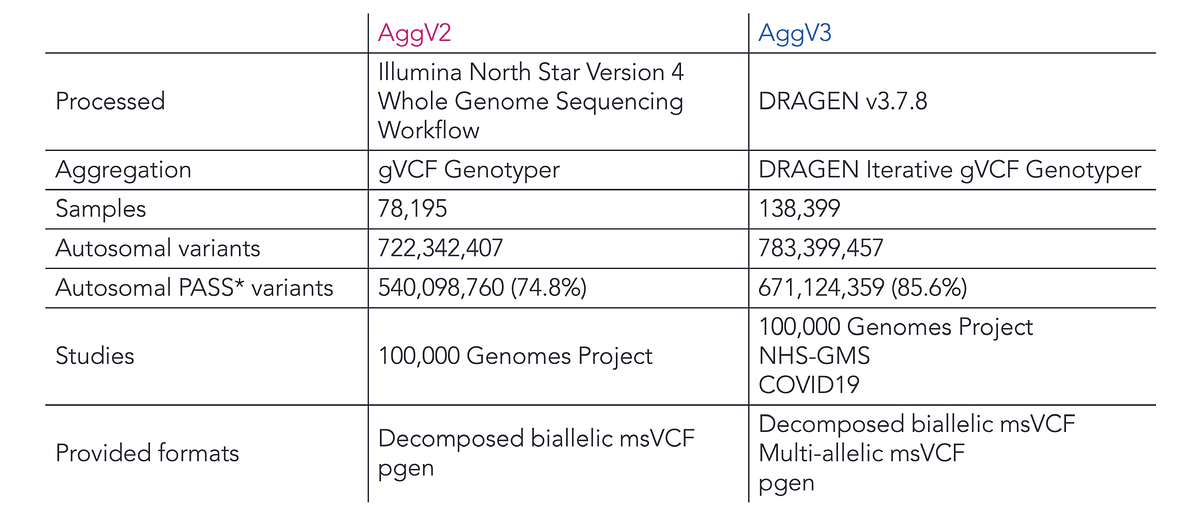
Table 1. Comparison between Genomics England previous variant aggregate and the new version developed with DRAGEN.
To illustrate the potential of the aggregates, Snow shared a story of how researchers used the previous variant aggregate from Genomics England to find a genetic association between a rare neurodevelopmental disease and a non-coding gene called RNU4-2. From there, researchers continued to investigate RNU syndromes and identified more disease-associated discoveries in this family of non-coding genes.
“What’s really nice about this is that we can now feed that back through to healthcare, so for anyone that comes along with a neurodevelopmental disorder, [physicians] will ensure that variants in those noncoding genes will now be prioritized through our interpretation pipelines,” said Snow. Next, her team plans to use DRAGEN to standardize their cancer data and will continue to use the tool to standardize incoming genomes in the future.
THE DRAGEN DIFFERENCE
Schulz-Trieglaff continued the webinar by discussing DRAGEN’s capabilities and what makes it stand apart from other tools. He highlighted its functionality, scalability, accuracy, input and output file formats, cost efficiency, and availability to sync with Illumina® Connected Analytics (ICA) and HPC environments.
In the context of variant aggregation at scale, the topic of joint genotyping comes up frequently and is often recommended to improve variant quality at the cohort level, Schulz-Trieglaff said. Joint genotyping is a mathematical algorithm popularized by the GATK software developed by the Broad Institute that calls variants across an entire cohort at once, using aggregate information from all samples to improve variant calling sensitivity in each individual sample. Schulz-Trieglaff emphasized that DRAGEN does not do joint genotyping in the same way as GATK joint genotyping (see Figure 1).
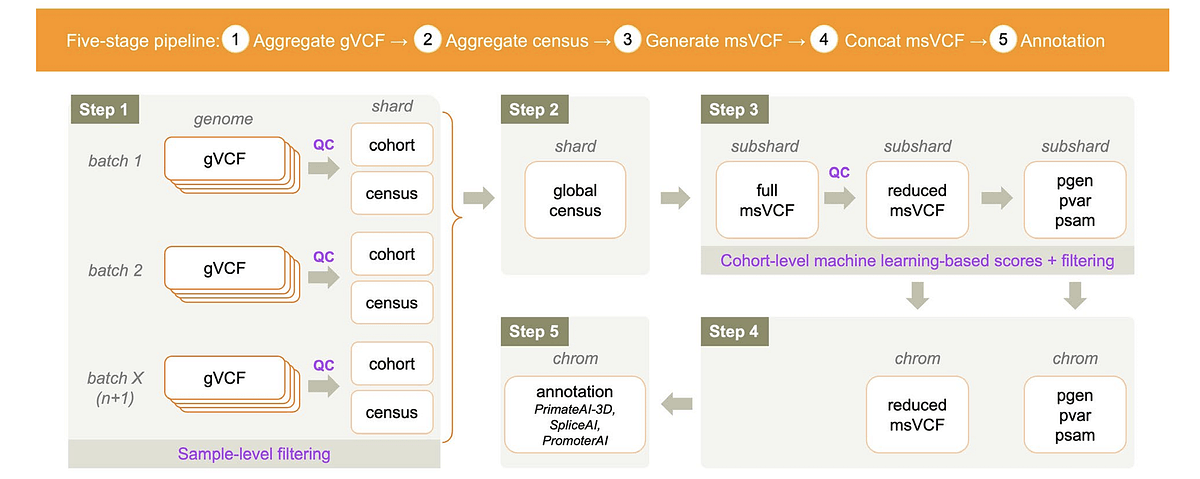
Figure 1. DRAGEN joint analyses in Illumina Connected Analytics.
“For unrelated samples that have been sequenced at high coverage, we do not recommend GATK joint genotyping,” said Schulz-Trieglaff. In his presentation, he shared data indicating an increased number of false positives and negatives generated for single-nucleotide polymorphisms and insertions and deletions using GATK joint genotyping (see Figure 2). In some cases, GATK joint genotyping was even detrimental to accurate variant aggregation when applied to germline variants generated by DRAGEN.
“The accuracy of DRAGEN is already very high,” said Schulz-Trieglaff. “What the GATK joint genotyping does is it tries to rectify errors in the call set that are not present because DRAGEN has already filtered them out 4 upstream.” He emphasized that GATK joint genotyping is expensive and not necessary with DRAGEN.
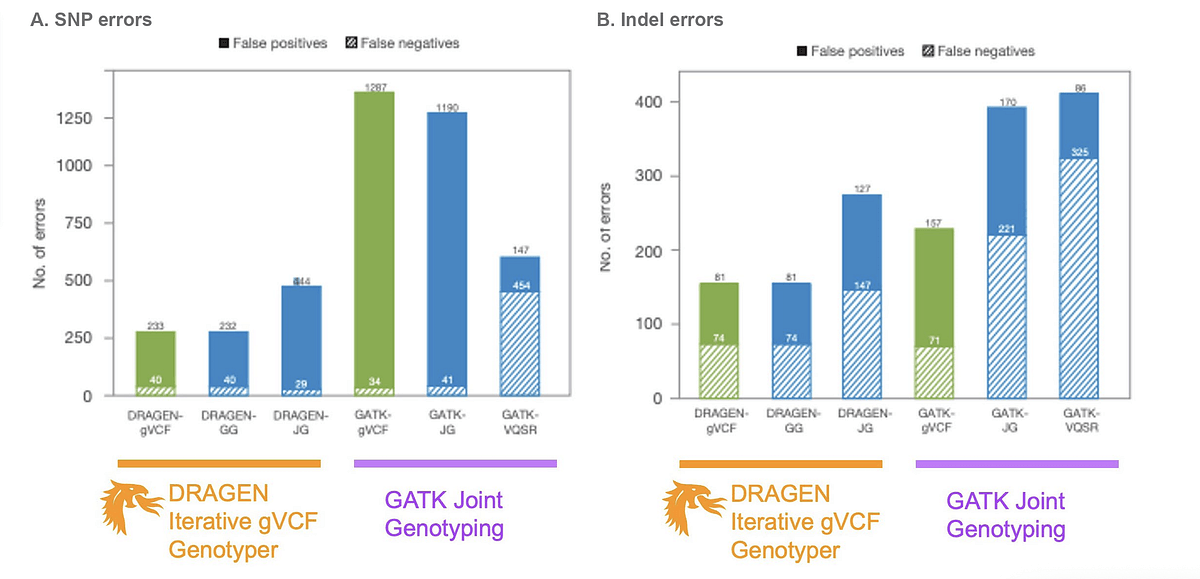
Figure 2. DRAGEN Iterative gVCF Genotyper vs. GATK Joint Genotyping
Next, Huang described DRAGEN’s distributed workflow. To effectively scale variant aggregation for large cohort studies, it splits sequenced genomes into regions and processes them in parallel. Broadly, the software combines numerous single-sample genomic variant call files (gVCFs) in a large, multi-variant dataset by processing samples in batches, summarizing their variant information and iteratively updating the cohort as new samples are added without redoing everything from scratch.
DETECTING THE RAREST VARIANTS
Huang highlighted Illumina’s most recent partnership with the UK Biobank and multiple biopharmaceutical companies to analyze approximately half a million whole-genome sequences using DRAGEN. The study results were recently published in Nature. “This is the largest-scale whole-genome sequencing effort and the first time we delivered at a cohort level almost half a million whole-genome variant aggregation results,” said Huang.
In his slides, he highlighted striking data that showed the statistical power and benefit of adding increasingly more samples and variant aggregation to detect ultra-rare variants that can appear in as few as just one person (see Figure 3). “You keep getting rare variants, which are extremely useful and important. That justifies why we want to do very large-scale variant aggregation and sequencing projects,” said Huang.
The project was completed using DRAGEN version 3.7.8, which already detects single-nucleotide polymorphisms, insertions, and deletions with high accuracy. However, Huang shared that, with machine learning improvements, they further increased the accuracy of DRAGEN version 3.7.8. The datasets from both analyses are publicly available online from the UK Biobank. The whole analysis was completed in Illumina Connected Analytics, a cloud environment provided by Illumina.
The Illumina team continues to improve DRAGEN. In the last portion of the webinar, Schulz-Trieglaff discussed the updates and features available on the latest version of DRAGEN including improvements to the identification of single-nucleotide variants, copy number variants, structural variants, and short tandem repeats. The research team at Illumina has also made improvements in variant annotation and in detecting variants in medically relevant genes, as well as HLA and PGx genes.
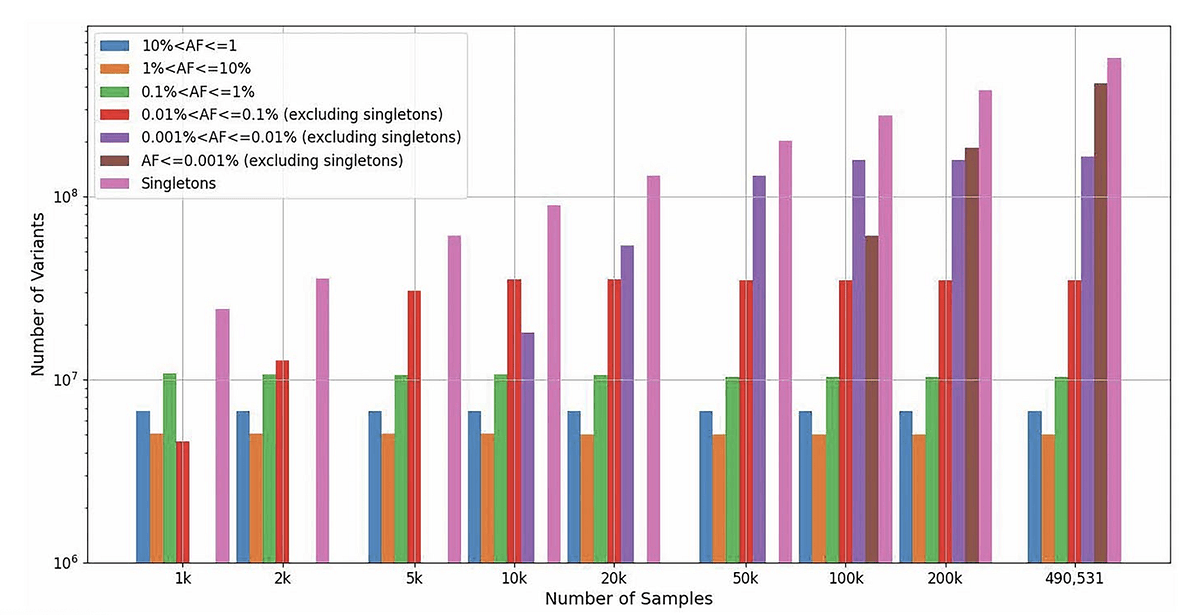
Figure 3. Number of variants in UK Biobank DRAGEN aggregated variant dataset in different allele frequency ranges as the number of samples increases from 1,000 to 490,541 (based on random downsampling). Variant alleles are collected from all autosomes, sex chromosomes, mitochondria, and ALT contigs. The UK Biobank Whole-Genome Sequencing Consortium. Whole-genome sequencing of 490,640 UK Biobank participants. Nature 645, 692–701 (2025). https://doi.org/10.1038/s41586-025-09272-9. CC BY 4.0
Schulz-Trieglaff also discussed how DRAGEN integrates with Illumina Connected Analytics, hosted on AWS cloud, for large-scale genomics. “What is important to realize is that association analysis is not a single monolithic tool, but it’s usually a workflow of different tools that are applied,” said Schulz-Trieglaff.
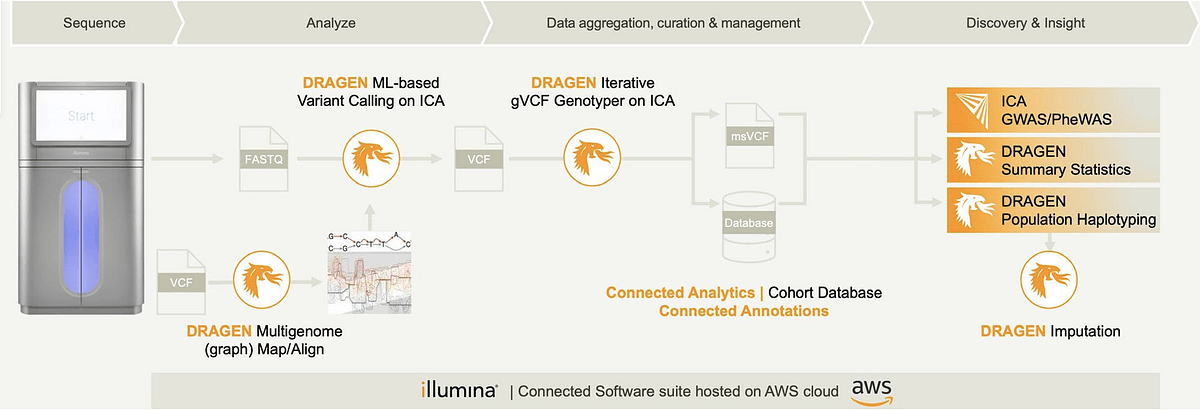
Figure 4. DRAGEN analysis (mapping/calling), Iterative gVCF Genotyper (aggregation), and Illumina Connected Analytics create a single, supported path from FASTQ to msVCF and statistical analysis — reducing integration risk and ensuring reproducibility.
Typical steps implemented in a GWAS analysis include ancestry analysis to assess cohort homogeneity and to account for differences in allele frequencies between different ethnicities, but also an analysis of sample relatedness and the detection of duplicate samples. “All of this is enabled using the highly accurate and filtered aggregate produced by DRAGEN Iterative gVCF Genotyper,” said Schulz-Trieglaff.

For Research Use Only. Not for use in diagnostic procedures.
M-GL-04242