- Home
- News & Updates
- Scalable Mosaic Variant Detection at Low Allele Fractions with DRAGEN
-
DRAGEN
-
News
- 03/19/2026
Scalable Mosaic Variant Detection at Low Allele Fractions with DRAGEN

Detecting mosaic variants below 5% variant allele fraction (VAF) at genome scale has remained largely impractical, until now. A recent medRxiv preprint1 introduces the DRAGEN mosaic variant caller, a hardware accelerated approach that enables sensitive, genome-wide detection of mosaic single nucleotide variants (SNV) and indels down to ~1-2% VAF, without requiring matched controls. This capability opens the door to population-scale and tissue resolved mosaic analyses that were previously out of reach.
Mosaic variants are post‑zygotic somatic mutations that arise in healthy tissues and are often present in only a small fraction of sequencing reads. Because these variants can clonally expand over time, they have been implicated in a wide range of conditions, including cancer initiation, neuropsychiatric disorders, and age-related pathologies, often emerging years or even decades before clinical symptoms appear.
At VAFs below 5%, true biological signal is easily obscured by technical noise, alignment artifacts, and sequencing errors. Progress has been further constrained by the lack of standardized benchmarks and the computational burden of scaling analyses across large datasets. As a result, low-VAF mosaic variation has largely escaped systematic study, despite its growing biological relevance across health, aging, and disease.
The preprint Scalable and comprehensive mosaic variant calling using DRAGEN directly addresses these challenges by introducing a low‑VAF–optimized mosaic variant caller within the DRAGEN framework, alongside a scalable, publicly available genome‑wide benchmark for evaluating mosaic detection across the 1–10% VAF range.
Discriminating true low-VAF mosaic signal from noise
Low allelic fraction makes mosaic variants particularly difficult to detect. Variants at 1–5% VAF fall within the error regime of short-read sequencing, where systematic noise and alignment artifacts can obscure true biological signal. Most small variant‑ callers are optimized to detect germline variants, where allele fractions cluster near 50% or 100%, and are not designed to reliably resolve low ‑fraction events.
To address these challenges, the preprint introduces the DRAGEN mosaic variant detection as an extension of the DRAGEN unified analysis framework (See Figure 1). Central to the approach is a dedicated machine learning model optimized for low VAF signal and integrated directly into DRAGEN’s hardware accelerated pipeline2.
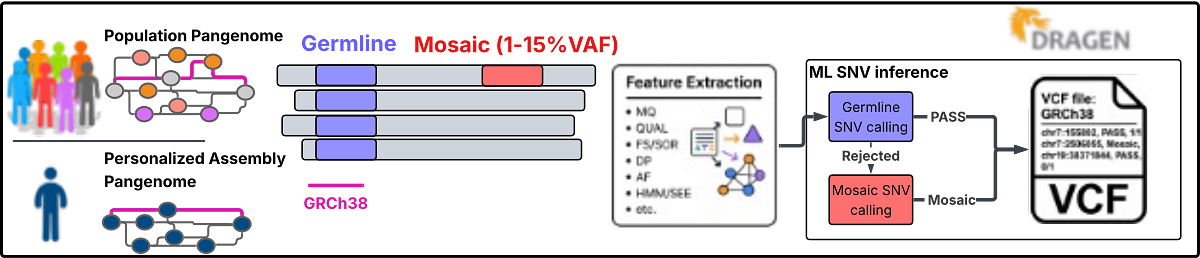
Figure 1: Overview of the DRAGEN mosaic variant detection approach, in which low-VAF (1–15%) candidate SNVs are processed through feature extraction and ML-based inference alongside regular germline calling.
The DRAGEN mosaic variant detection, notably, does not require a matched control sample to call mosaic variants from a sample’s tissue, offering the unique advantage of reporting mosaic variants alongside germline calls within a single, unified workflow, with calls discriminated by dedicated tags in the VCF output. All complemented by a minimal runtime overhead in addition to the standard DRAGEN germline variant caller pipeline, enabling streamlined, high-throughput analyses without sacrificing performance. Further, the preprint presents a companion high-sensitivity mode, that allows to increase sensitivity in the lower VAF range, despite a minor increase in runtime and false-positive rate.
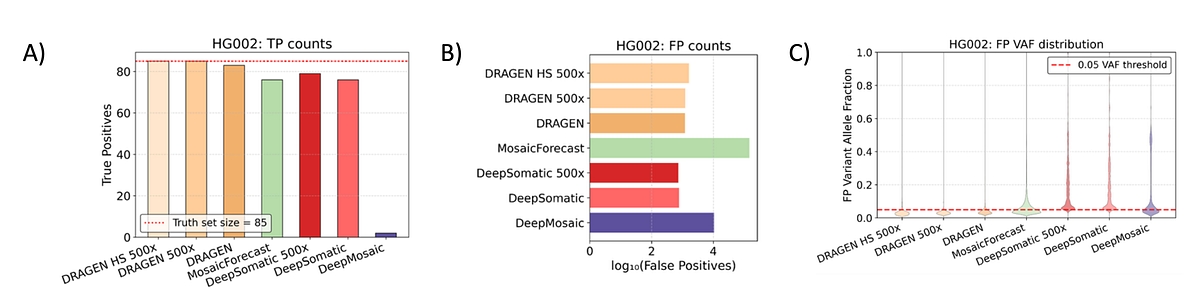
Figure 2: A) True positives (TP) counts for HG002 GIABv1.1 Mosaic benchmark; DRAGEN detected all 85 variants at 500× coverage in both regular and high-sensitivity (HS) mode and missed two at 80× in regular mode due to downsampling issues. DeepMosaic detected 84/85 variants but labeled 82 as artifacts, correctly annotating only two. B) False positives (FP) counts for HG002 GIABv1.1 Mosaic benchmark; MosaicForecast produced the highest false‑positive count (146,616), followed by DeepMosaic, while DRAGEN showed substantially fewer false positives. DeepSomatic had the lowest absolute false‑positive count. C) True positives (TP) variant allele fraction distribution for HG002 GIABv1.1 Mosaic benchmark; A large fraction of MosaicForecast (46.2%) and DeepMosaic (59.1%) false positives were above the 5% VAF threshold, whereas DRAGEN had the lowest proportion above threshold (15.8%), with only 4.3% at higher coverage. DeepSomatic false positives were all above 5% VAF.
We evaluated the DRAGEN mosaic caller sensitivity against recently established benchmarks. In direct comparisons with established mosaic and somatic variant callers including DeepMosaic3, MosaicForecast4, and DeepSomatic5, the DRAGEN mosaic variant caller consistently identified more true mosaic variants (See Figure 2A) while reporting fewer false positives (See Figure 2B) across the low-VAF regime (See Figure 2C). On the Genome in a Bottle HG0026 benchmark (85 high confidence mosaic SNVs over 2.45 Gbp of benchmark regions), DRAGEN achieved complete detection at 500× coverage while maintaining controlled false positive rates within the benchmarked 5–30% VAF range. DeepSomatic detected only a subset of the true positive variants at the same depth. To allow a comparison with the other callers, the same dataset was downsampled at 80x. DRAGEN missed two variants due to down-sampling related issues. Other callers detected fewer true positives (76-2 TPs when disregarding filters) with higher false positives (768-67,729 FPs when considering filters) in the 5-30% VAF range. Notably, DRAGEN analyzed the 500x sample from alignment to variant calling in little above 12 hours, with other methods running 4-7x slower than DRAGEN in the end-to-end setting.
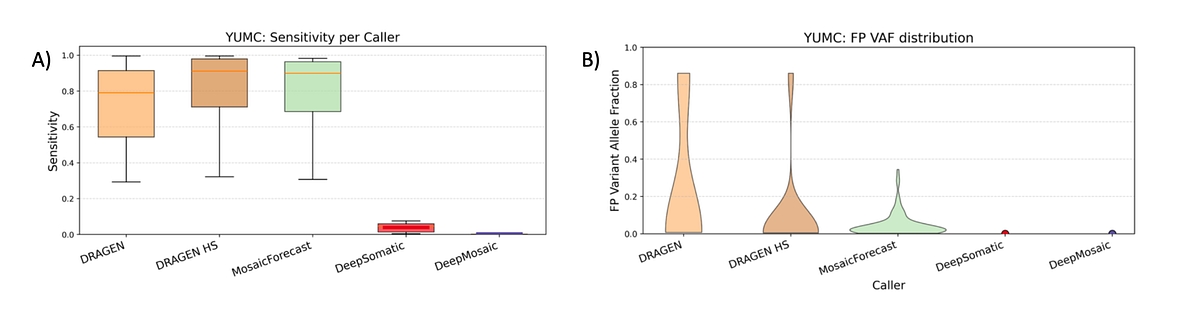
Figure 3: A) Sensitivity results for YUMC benchmark. Per-cell mixture 2,316-9,549 true-positive variants are detectable, with similar patterns across DRAGEN and MosaicForecast, while DeepSomatic detects only up-to 545 TPs and DeepMosaic classifies almost all calls as artifacts B) False positive VAF distribution for YUMC benchmark. Only MosaicForecast has significant numbers of false positives; however, 54% of those variants are under the 5% VAF mosaic detection threshold. DeepSomatic reports 2 FP variants, whereas DRAGEN showed 13 variants across 8 cell mixtures.
We also evaluated the Yonsei University College of Medicine (YUCM) mosaic benchmark7, a collection of in-vitro cell-line admixtures at different concentrations obtained to represent a variety of VAFs. DRAGEN preserved high average recall with 71.4% and 81.2% for regular and high-sensitivity modes, respectively (See Figure 3A). MosaicForecast showed an average recall of 69.1% while DeepMosaic reported an average recall of 69.1%. DeepSomatic reported the least number of true positives with an average recall of 4%. All methods reported very few false positives with DRAGEN, MosaicForecast, and DeepMosaic all reporting precision >99.4% with MosaicForecast being the least accurate and DeepSomatic being more accurate (See Figure 3B).
Beyond synthetic and reference datasets, the study applies the DRAGEN mosaic caller to real biological samples, including blood, brain, and sperm, demonstrating applicability across diverse tissue contexts.
Addressing evaluation gaps and genomic complexity in low‑VAF mosaic detection
Progress in low‑VAF mosaic variant detection has also been limited by the lack of comprehensive genome‑wide benchmarks. While germline variant calling has benefited from well-established reference datasets, mosaic benchmarking resources remain sparse and limited in scope. Existing resources capture only partial aspects of mosaic variation: the YUCM benchmark is restricted to deeply sequenced exomes and excludes complex genomic regions, while Genome in a Bottle HG002 is genome‑wide but reports only a small number of SNVs at VAFs ≥5%, omitting indels and lower‑fraction variants.
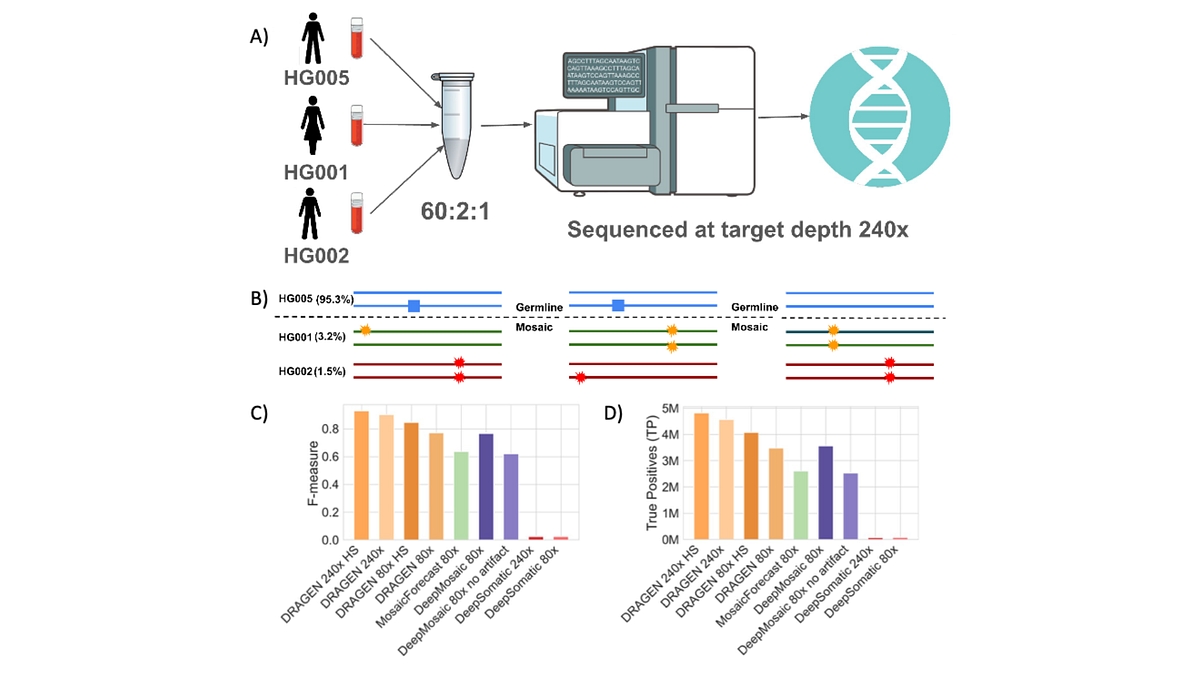
Figure 4: A) Three GIAB samples HG005, HG001, and HG002 were mixed in-vitro with 60:2:1 ratio and sequenced at 240x coverage B) Mosaic variants based on their presence in HG001 and HG002 C) Comparison of TP of different methods D) Comparison of F-measure. DRAGEN was run with both default and high-sensitivity (HS) mosaic detection mode; on both 240x ad 80x downsampled dataset.
To address this gap, the preprint introduces a scalable benchmark derived from mixtures of well‑characterized reference samples from GIAB, spanning variants from approximately 1–10% VAF across the genome. This HapMap low‑VAF benchmark provides the first genome‑wide resource for evaluating variant callers in a VAF range that has remained largely inaccessible due to the absence of orthogonal truth sets (See Figure 4). By bridging germline and mosaic analyses, it enables standardized, quantitative comparison of low‑fraction variant detection across methods and technologies. At 240x DRAGEN achieved an F‑measure of 90.14% (82.15% sensitivity, 99.84% precision), which improved to 92.78% F‑measure and 86.78% sensitivity using high‑sensitivity mosaic mode with minimal precision loss. When stratified by VAF, DRAGEN achieved >95% recall for variants at 3% VAF and above, 74.4% recall for variants with VAF between 3% and 1.5%, and 34% recall for variants with VAF below 1.5%, mostly limited due to insufficient ALT supporting coverage. When downsampled at 80x DRAGEN overall recall drops to 62% and 73% in regular and high-sensitivity mode, respectively. DeepMosaic recall is 64% while having two orders of magnitude more false positives than DRAGEN, MosaicForecast recall is 45% with one order of magnitude more false positives than DRAGEN, with DeepSomatic detecting the lowest number of true positives at both 240x and 80x.
In parallel, the work introduces personalized assembly pangenome references (PAPR) as a complementary advance aimed at improving accuracy in the same challenging genomic contexts highlighted by these benchmarks. By incorporating phased diploid assemblies into DRAGEN’s pangenome framework, this approach improves variant calling in structurally complex and polymorphic regions that are poorly represented by a single linear reference, while preserving GRCh38 coordinates for downstream compatibility. Benchmarking demonstrates consistent gains in germline precision and recall, and application to real tissues helps clarify tissue‑specific mosaic patterns while distinguishing in vivo mosaic variation from cell‑culture artifacts.
Looking ahead: from background noise to biological signal
Together, these advances mark a turning point in mosaic variant analysis, establishing DRAGEN as a powerful platform for making low‑VAF mosaic variation measurable at scale. By combining low‑VAF‑optimized modeling, realistic benchmarking, hardware‑accelerated performance, and pangenome‑aware alignment, DRAGEN enables population‑scale and tissue‑resolved mosaic studies that were previously impractical. Across multiple evaluations, the method demonstrated strong sensitivity for mosaic SNVs and indels at approximately 2–3% VAF, with partial sensitivity extending toward 1% given sufficient coverage, while maintaining low false positive.
As sequencing datasets continue to grow, the ability to routinely interrogate low‑VAF mosaic variation will enable deeper insights into clonal dynamics, tissue heterogeneity, and early molecular signatures of disease. Mosaic variation is no longer an edge case; it is a fundamental layer of genome biology that can now be studied systematically across health, aging, and disease.
For those interested in reading the technical specifics, please read the full preprint on medRxiv.
M-GL-04186
References
- Behera, S., Rossi M., Wang, Y., Izydorczyk, M.B., et al. (2026). “Scalable and comprehensive mosaic variant calling using DRAGEN.” medRxiv. https://doi.org/10.64898/2026.02.03.26345450
- Behera, S., Catreux, S., Rossi, M. et al. Comprehensive genome analysis and variant detection at scale using DRAGEN. Nat Biotechnol 43, 1177–1191 (2025). https://doi.org/10.1038/s41587-024-02382-1
- Yang, X., Xu, X., Breuss, M. W., et al. (2023). Control‑independent mosaic single nucleotide variant detection with DeepMosaic. Nat Biotechnol, 41, 870–877. https://doi.org/10.1038/s41587-022-01559-w
- Dou, Y., Kwon, M., Rodin, R. E., et al. (2020). Accurate detection of mosaic variants in sequencing data without matched controls. Nat Biotechnol, 38, 314–319. https://doi.org/10.1038/s41587-019-0368-8
- Park, J., Cook, D., Chang, P.‑C., et al. (2025). Accurate somatic small variant discovery for multiple sequencing technologies with DeepSomatic. Nat Biotechnol, 1–10. https://doi.org/10.1038/s41587-025-02839-x
- Daniels, C. A., et al. Characterization of subclonal variants in HG002 Genome in a Bottle reference material as a resource for benchmarking variant callers. Cell Genomics, 0(0), 101104. https://doi.org/10.1016/j.xgen.2025.101104
- Ha, YJ., Kang, S., Kim, J. et al. Comprehensive benchmarking and guidelines of mosaic variant calling strategies. Nat Methods 20, 2058–2067 (2023). https://doi.org/10.1038/s41592-023-02043-2