- Home
- News & Updates
- Somatic Pipeline Improvements with DRAGEN v3.3
-
DRAGEN
-
Product updates
- 05/29/2019
Somatic Pipeline Improvements with DRAGEN v3.3
Significant accuracy gains and speed improvements with DRAGEN v3.3, released April 2019
by Severine Catreux - Associate Director, Bioinformatics FPGA Development
The DRAGEN engineering and bioinformatics team is excited to announce a new DRAGEN release, v3.3. The second of several releases scheduled for 2019, DRAGEN v3.3 contains improvements across the many pipeline offerings now supported by the DRAGEN platform. This includes accuracy improvements in the germline and somatic pipelines, new features (e.g. CNV DeNovo calling and RNA quantification) and speed gains (Somatic T/N, BCL conversion).
Please see DRAGEN v3.3 Release Notes for more details. This blog highlights the significant updates to the DRAGEN Somatic Pipeline for small variants, that are part of the v3.3 release.
As one of DRAGEN’s core pipelines, the DRAGEN Somatic Pipeline for small variants is utilized by cancer research institutes around the globe. Expanding on the existing functionality, accuracy and speed of the DRAGEN Somatic Pipeline, the v3.3 release placed a high focus on the somatic tumor/normal WGS mode, producing step-function improvements in both accuracy and speed.
Accuracy Improvements:
During the development cycle for v3.3, the DRAGEN engineering and bioinformatics teams took a deep dive into the DRAGEN Somatic Pipeline tumor/normal mode, strengthening the existing algorithm for accuracy improvements. Specific improvements were made in the genotyping module, to replace point estimation of the variant allele frequency with continuous integration over a range of possible frequencies. This led to significant gains in both sensitivity and precision. Additionally, downstream filtering rules were improved to optimize both sensitivity and precision (less stringency on clustered variants, filter variants positioned at the edge of reads, filter variants with low median base quality and MAPQ). Finally, the indel PCR error model autocalibration module was made independent between the tumor and normal control, to allow for differences in library preparation between the tumor sample and the control sample.
These changes are precursors to further accuracy improvements planned for the DRAGEN v3.4 release, specifically in the area of liquid tumor support, where tumor-in-normal contamination will be taken into account.
Accuracy gains of DRAGEN 3.3 over previous DRAGEN versions (3.2) as well as other pipelines (GATK4 MuTect2 and Strelka2) are shown in the plot below. Gains are measured for both SNVs and indels on most datasets.
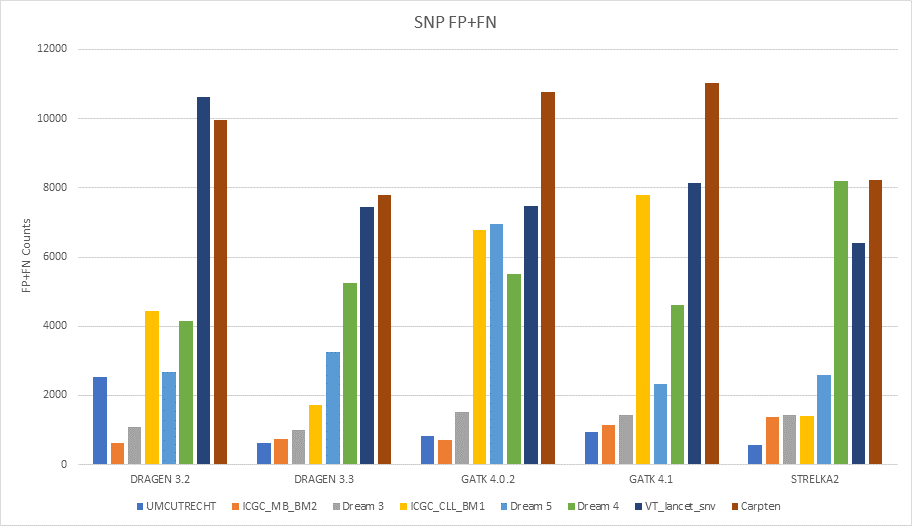
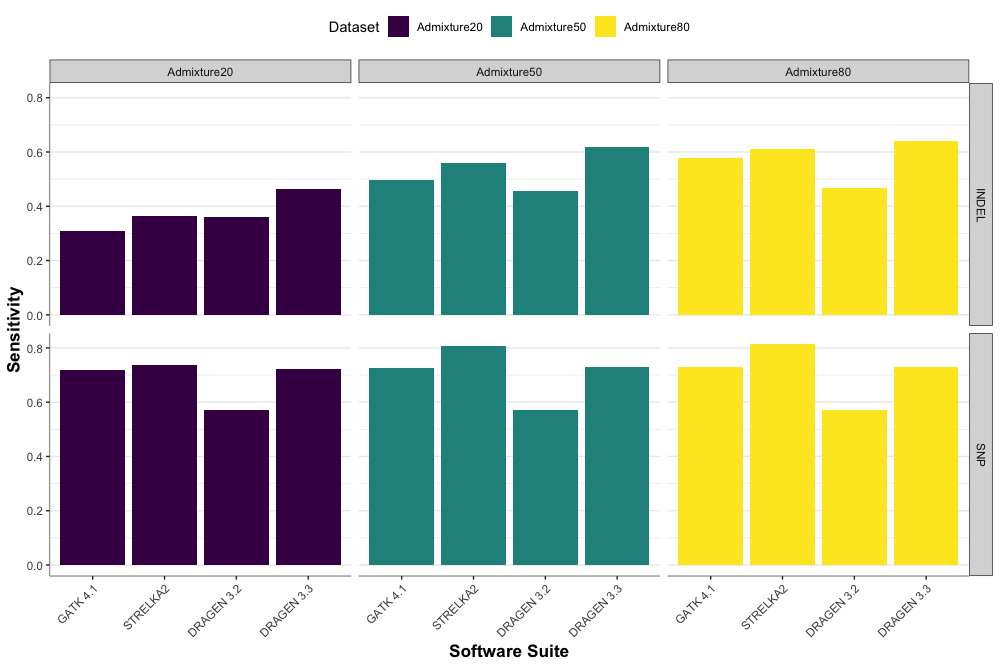
Speed Gains
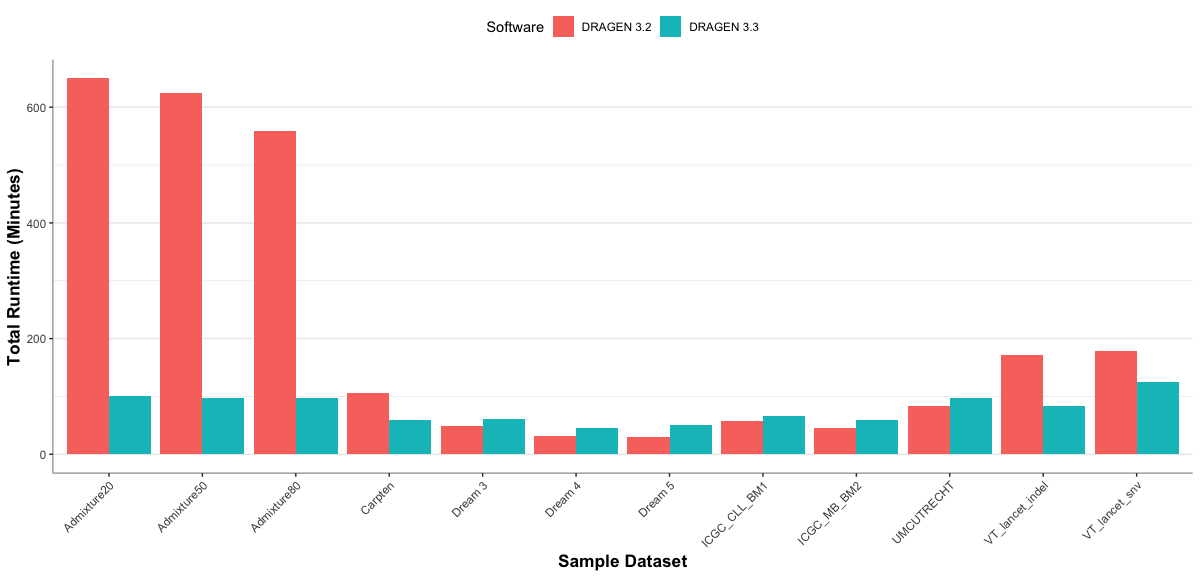
DRAGEN v3.3 delivers unprecedented fast run times on the processing of somatic T/N WGS. Users of previous DRAGEN versions will notice substantial speed gains in DRAGEN 3.3 (see graph below). For datasets that were previously HMM-limited, v3.3 delivers up to 6-fold speed improvements, with a typical 100x (tumor) and 40x (normal) run finishing within 1 hour and 40 minutes on an on-premise DRAGEN server. In the cloud, run times average at 2 hours and 30 minutes.
The run time gains were obtained from optimizations in the upstream stages of the pipeline (more efficient way of defining regions of interest and increase the MAPQ threshold of reads to pass downstream, i.e., less reads get passed downstream, without loss on sensitivity). Additionally, the accelerated HMM engines were optimized to consume less of the FPGA footprint, such that more engines could be run in parallel.
Run-time comparison for T/N WGS Somatic Calling
About the DRAGEN Somatic Pipeline
The DRAGEN Somatic Pipeline provides highly accurate, ultra-rapid secondary analysis for tumor-only and tumor/normal experiments to identify cancer-associated mutations.
Tumor/Normal Mode
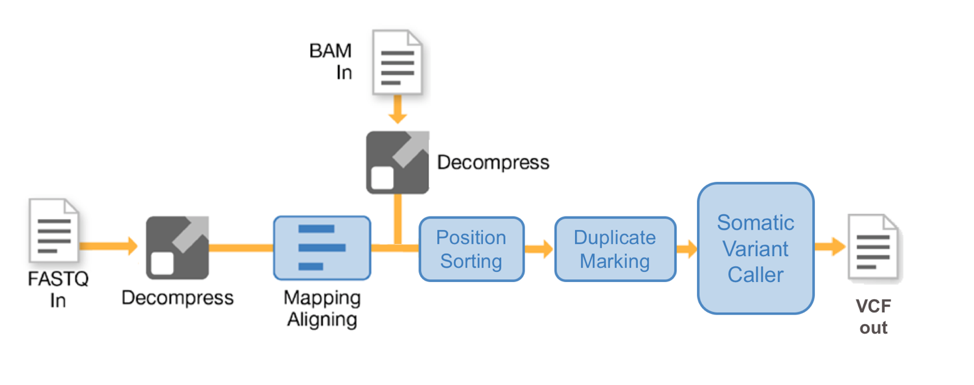
The DRAGEN Somatic Pipeline offers flexible data analysis to suit the specific needs of users. DRAGEN accepts FASTQ, BAM/CRAM, and BCL files and supports NGS input from whole genome, whole exome, and targeted cancer panels. In the tumor/normal pipeline, both samples go through identical processing steps of mapping, aligning, sorting, and duplicate marking. Then, both sets of tumor and normal reads are passed through the somatic variant caller which looks for sites exhibiting a mutation in the tumor reads while showing little to no evidence of the mutation in the normal reads, thus producing a VCF file containing tumor-specific mutations. The Somatic Pipeline also reports allele frequency, allowing users to assess the prevalence of a specific mutation.
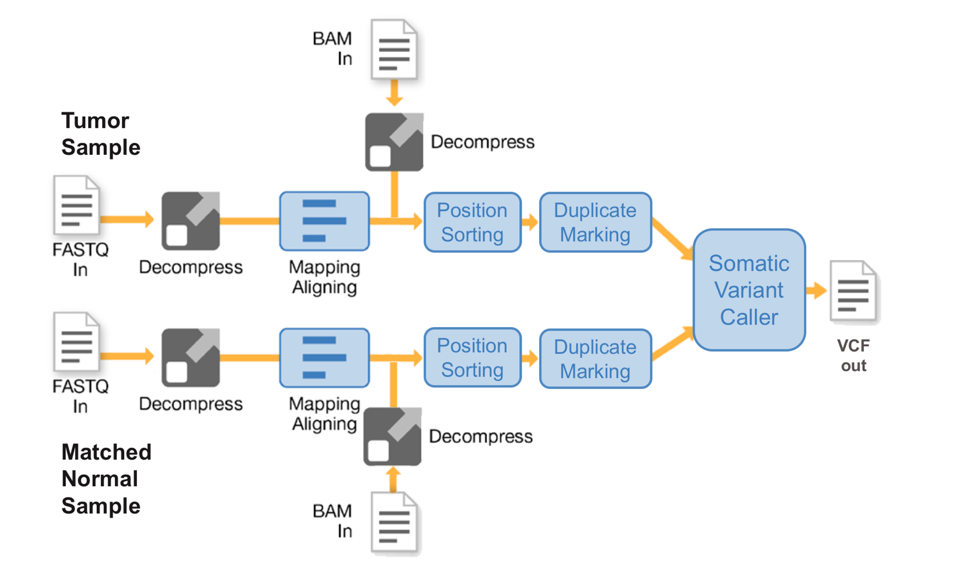
Tumor-only Mode
In the tumor-only pipeline, users input NGS data from a tumor sample and run it through the same pipeline as for tumor/normal analysis, but it lacks the matching normal sample. The somatic variant caller contains algorithms that distinguish low-frequency alleles from background noise. Although the resulting VCF file does not distinguish germline from somatic variants, it allows researchers and clinicians to determine if a mutation is present in a tumor sample and its allele frequency.
Have any feedback, suggestions or data that you’d like to share with the DRAGEN team? Our new community forum is an active, collaborative hub for connecting and sharing feedback.
For Research Use Only. Not for use in diagnostic procedures