- Home
- News & Updates
- Frequently Asked Questions - DRAGEN v4.5™: Expanding Accurate Variant Discovery Across the Entire Genome
-
DRAGEN
-
Product updates
- 06/22/2026
Frequently Asked Questions - DRAGEN v4.5™: Expanding Accurate Variant Discovery Across the Entire Genome

On April 30, 2026, Illumina hosted a technical webinar introducing DRAGEN v4.5, one of the most substantial expansions of the software's capabilities to date. This release delivers significant advances spanning machine learning (ML)–driven secondary analysis to improved somatic variant detection for FFPE and low VAF samples, enabling researchers to unlock deeper biological insights while maintaining production-scale performance.
Following the webinar, our Illumina experts compiled this FAQ resource to address audience questions from the two sessions of DRAGEN v4.5: Expanding Accurate Variant Discovery Across the Entire Genome.
Frequently Asked Questions
01 — Overview of DRAGEN Secondary Analysis
Q: How does DRAGEN v4.5 work with the hg19 reference genome?
A: DRAGEN supports hg38, hg19, and hs37d5/GRCh37.
Q: Is DRAGEN v4.5 available to use on AWS?
A: Yes, DRAGEN v4.5 is available via Bring Your Own License (BYOL) or marketplace offering. Contact Tech Support for access to a private AMI, or subscribe directly through AWS marketplace.
Q: Does iCredits pricing change with v4.5? Are any apps still free?
A: No changes to any commercial model. Illumina® Connected Analytics (ICA) apps continue to use iCredits as before, and any apps previously marked free will remain free.
Q: Can DRAGEN v4.5 be used on-premises servers?
A: Currently we are referring to DRAGEN Servers with FPGAs.
Q: Is DRAGEN v4.5 enabled on sequencers like NovaSeq™ X?
A: Not currently. If sequencer support is added in the future, it will be limited to upgrades of the existing pipelines only .
Q: Where can I find user guides and reference manuals to use DRAGEN v4.5 on my server?
A: Best practices documented in the DRAGEN Recipes: https://help.dragen.illumina.com/dragen-v4.5/product-guides/dragen-v4.5/dragen-recipes
For annotation, Nirvana supports a VCF output mode for a subset of annotations: https://help.connected.illumina.com/annotation/v3.27/file-formats/illumina-annotator-vcf-file-format
Q: Can DRAGEN v4.5 analyze data from non-Illumina short read sequencers?
A: No. DRAGEN is validated only on Illumina data. Illumina's EULA also prohibits use with data from other sequencing platforms.
02 — DRAGEN Germline
Q: How does DRAGEN v4.5 perform for SMA silent carrier detection across ancestral populations, and can the biomarker-based approach extend to other paralogs?
A: Overall recall is 90% based on the 1000 Genomes (1KG) cohort, though this figure is weighted toward African populations, which account for >50% SMN1 duplication events in the cohort. Performance is strongest in African populations, where 2-copy allele frequencies are higher, see Figure 1. In populations with low 2-copy allele frequencies, performance degrades - for example, East Asian samples represented only 2% of the duplication events (~25 events), none of which were detected (recall = 0%) with the current biomarker set.
Work is ongoing to incorporate additional biomarkers and expand the training cohort. Extension of this haplotype detection framework to other paralogs such as CYP21A2/CYP21A1P is feasible and under exploration.
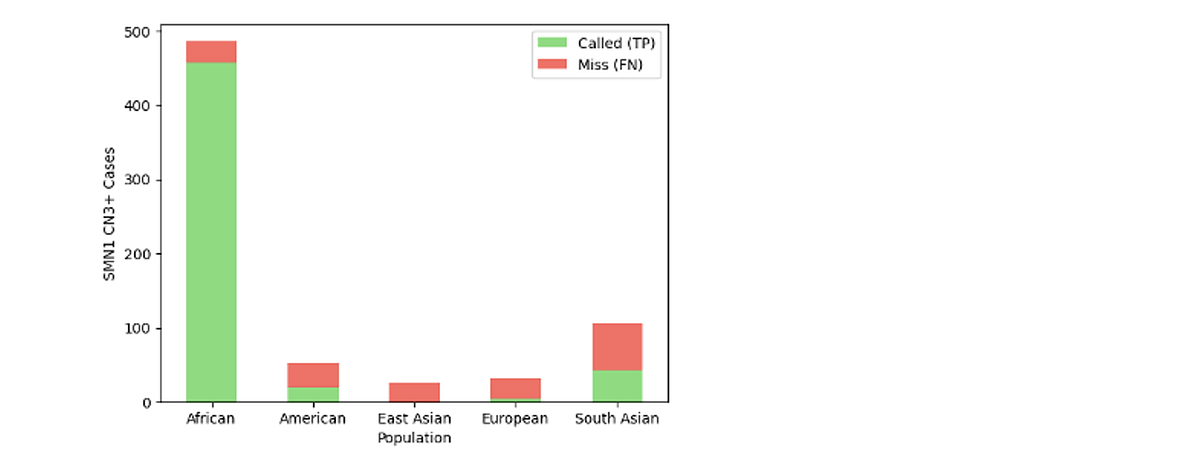
Figure 1.
Q: Does DRAGEN v4.5 SMN caller support long-read data such as PacBio HiFi ?
A: No, the DRAGEN v4.5 SMN1 caller is designed for short-read WGS data and relies on read depth and read count statistics specific to that context. It is only supported for PCR-free human WGS libraries.
Please note that Illumina's EULA also prohibits use with data from other sequencing platforms.
Q: How accurate is DRAGEN v4.5 for STR calling, including repeat number estimation?
A: Benchmark data for HG002 are available in the DRAGEN Nature Biotechnology paper (https://www.nature.com/articles/s41587-024-02382-1/figures/2), and performance data are reported in The Lancet Neurology (https://www.thelancet.com/journals/laneur/article/PIIS1474-4422(21)00462-2/fulltext). The TruPathTM Genome technical paper, forthcoming, will report substantially improved repeat number accuracy. Repeat number estimation uses a sequencing efficiency correction model derived from samples with orthogonal truth data. The IRR recovery model improves mapping of informative reads, and residual signal loss — which is approximately linear for loci such as DMPK — is estimated probabilistically.
Q: Can DRAGEN v4.5 detect mitochondrial CNVs?
A: Yes. Mitochondrial deletions and duplications (typically kilobase-scale events) are reported in the DRAGEN SV VCF. Smaller mitochondrial variants, including SNPs and indels and handled by the small variant caller.
Q: How does DRAGEN v4.5 calculate absence of heterozygosity (AOH), and what visualization options are available?
A: AOH analysis uses a population SNP catalog, downloadable for the appropriate reference from the DRAGEN resource page: https://support.illumina.com/sequencing/sequencing_software/dragen-bio-it-platform.html. Output files are compatible with IGV via standard session XML, bigwig, and bedgraph formats. For downstream interpretation, Emedgene and Illumina Connected Insights (ICI) also support CNV/AOH visualization. Detailed documentation on intermediate and visualization files is available at: https://help.dragen.illumina.com/dragen-v4.5/product-guides/dragen-v4.5/dragen-dna-pipeline/cnv-overview/cnv-reference#intermediate-and-visualization-files
Q: What's the limit of detection (LoD) for CNV calling in standard WGS using DRAGEN 4.5?
A: Benchmarked against the HG002 NISTv0.6 truthset, DRAGEN CNV achieves an F1 score of >97% for standard WGS coverage.
Q: What's the CNV detection sensitivity in low-pass WGS?
A: Compared to Chromosomal Microarray (CMA), lpWGS at 1-2x coverage detects events >=200kbp with >90% sensitivity. Analytical sensitivity increases substantially at 5-10x coverage and for events >=500kbp. Labs interested in working on a formal study with us to assess lpWGS as a replacement for legacy methods are encouraged to reach out to your Illumina representative.
Q: How does DRAGEN CNV normalization work in absence of a cohort?
A: Two normalization strategies are supported: self-normalization for WGS, which exploits the uniform coverage profile, and a Panel of Normals approach for WES and small panels to account for capture-related coverage biases. Full details are in the application-specific user guides: https://help.dragen.illumina.com/product-guides/dragen-v4.5/dragen-dna-pipeline/cnv-overview.
Q: What coverage range does DRAGEN v4.5’s low-pass CNV caller support, and can it detect mosaic events?
A: DRAGEN v4.5 low-pass CNV caller has been successfully tested down to 1x coverage. At 0.5x, the pipeline runs but accuracy claims are not yet supported. Collaborators interested in evaluating performance at these shallow depths are welcome to reach out to your Illumina representative.
Large-scale CNV events including whole-chromosome aneuploidy and mosaic CNVs are detectable in shallow data. The minimum detectable event size in low-pass mode is ~200 kbp; events smaller than 50 kbp are better addressed with standard ~30× WGS.
Q: How can SQ, MQ, and other quality scores be adjusted in DRAGEN v4.5?
A: Quality score configuration is described in the variant scoring section of the user guide. DRAGEN quality scores are derived from a probabilistic model incorporating mapping quality and sample-specific noise. When ML-based scoring is enabled, scores are further recalibrated using read-level and contextual features to better reflect true variant confidence.
You can find more details in the user guide for variant scoring.
DRAGEN variant quality scores (including SQ and MQ) are derived from a probabilistic model that incorporates multiple signals, such as mapping quality and sample-specific noise.
When ML–based scoring is enabled, DRAGEN further recalibrates quality scores using read-level and contextual information, improving how well scores reflect true variant confidence.
Q: Does DRAGEN v4.5 support HLA typing and what resolution is achievable?
A: HLA typing is supported for both WGS and WES 4-field resolution typing requires coverage of non-coding regions, hence supported for whole genome assays only. The plots featured in the webinar slide (see Figure 2) are from WGS captures the performance of HLA typing. WES supports for 2-3 field typing, with performance comparable to the 2-3 field results observed in WGS benchmarking in Figure 2.
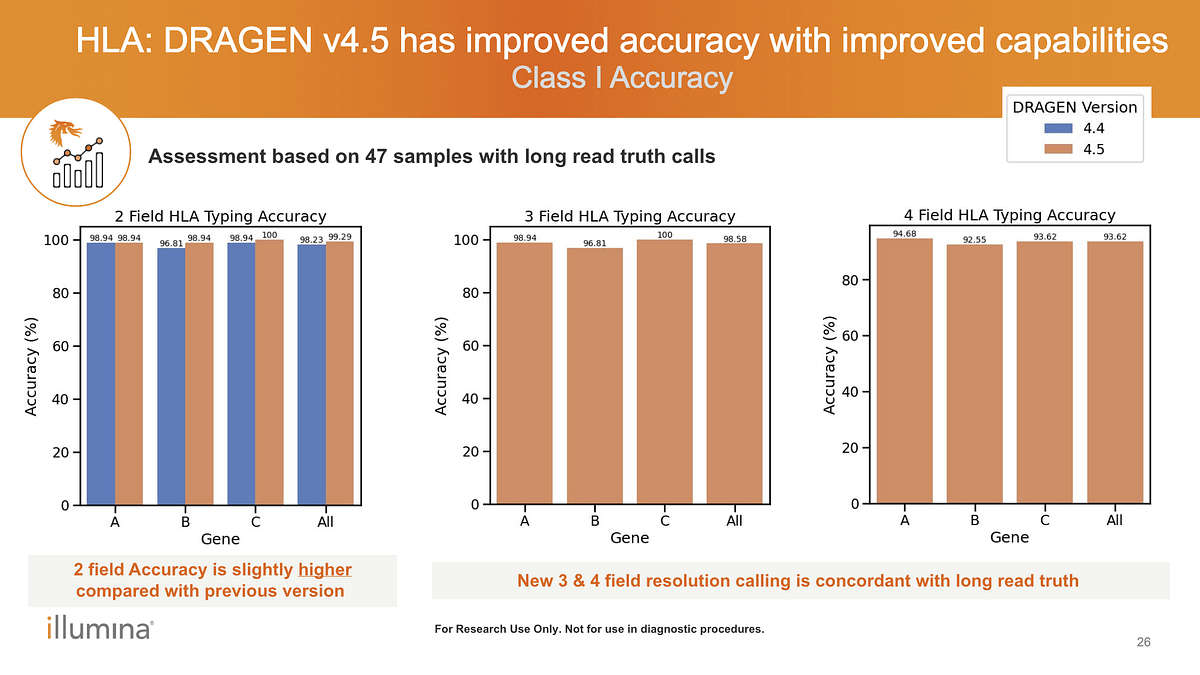
Figure 2. HLA typing performance of DRAGEN v4.5 across resolution levels.
WGS benchmarking against long-read truth (n=47 samples) shows high accuracy for 2-field, 3-field, and 4-field HLA typing, with improvements in higher-resolution calls.
03 — DRAGEN Somatic
Q: How does DRAGEN v4.5 handle SNVs calling differently for FFPE vs Fresh-frozen (FF) samples?
A: The ML model has been trained on both FFPE and FF data to handle SNV calling. As with the current non-ML workflow, no separate settings are required – the same settings can be applied to both types of sample preparation.

Q: When can we expect Somatic ML be production-ready?
A: Somatic ML is planned for production release in v4.7. Feedback on regressions and on prioritization of recall improvements vs. false positive reduction is welcome. The v4.7 release will also include a production-ready MRD application with QC, fingerprint tracking, longitudinal analysis, and MRD reporting.
Q: What data was used to benchmark Somatic ML for FFPE noise filtering?
A: Benchmarking was performed on WGS data, see Figure 3. ML can also be applied to WES. In this release, tumor/normal (T/N) paired analysis is required; tumor-only ML support is in development for the next release.
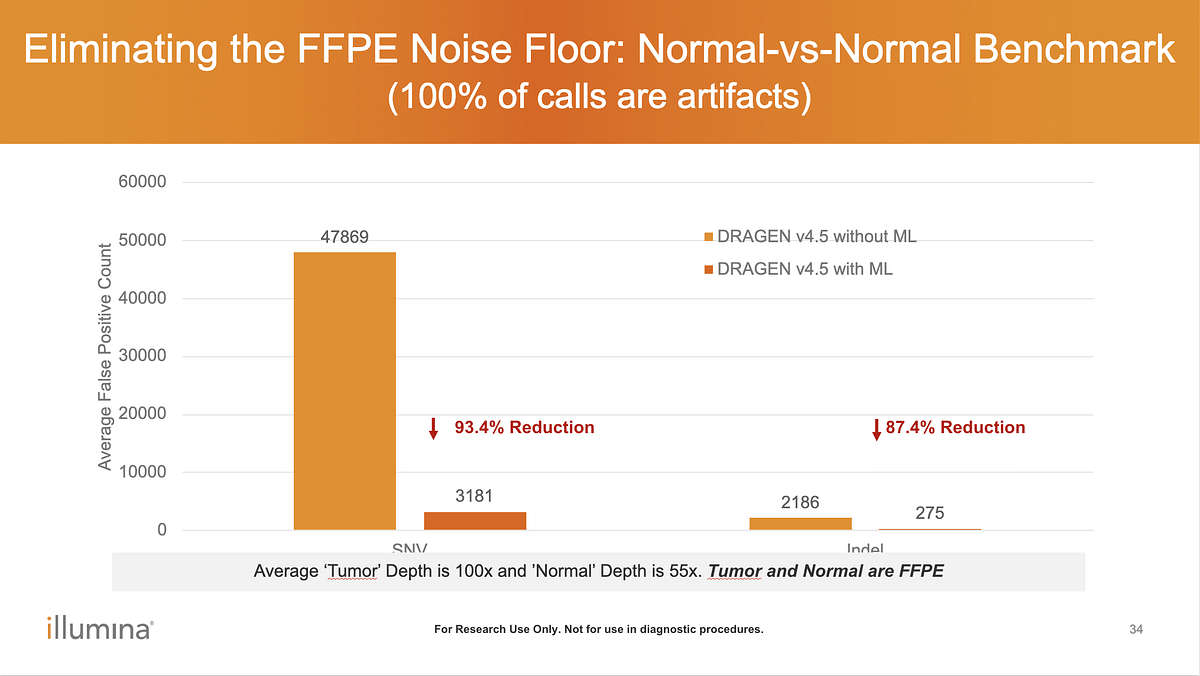
Figure 3. FFPE artifact reduction in normal–normal benchmarking using DRAGEN v4.5 Somatic ML.
Normal–normal WGS benchmarking (100% expected artifacts) demonstrates the impact of ML-based filtering on FFPE-induced noise. Enabling ML in DRAGEN v4.5 substantially reduces false-positive SNV and indel calls compared to non-ML mode (average tumor depth 100×, normal depth 55×; both FFPE).
Q: Is tumor-only somatic calling supported in DRAGEN v4.5’s ML support?
A: Not in this release. /T/N paired analysis is required. tumor-only ML support is in development for the next release.
Q: How does DRAGEN handle samples with low viral load in oncovirus detection, and can the virus database be customized?
A: 1) For low viral load samples, the --oncovirus-detection-below-threshold flag can be enabled. The current oncovirus database covers 7 oncoviruses including 25+ HPV types and is not user-configurable. To add custom sequences, contact Illumina support. Database details are in the user guide: https://help.dragen.illumina.com/dragen-v4.5/product-guides/dragen-v4.5/dragen-dna-pipeline/oncovirus-detection
Q: What minimum coverage is recommended for accurate small variant calling (SNVs + indels)?
A: No change from previous versions. DRAGEN requires a minimum of 3 alt-supporting reads per call. At 100× mean coverage, variants down to ~3% VAF are detectable in principle, though site-level coverage variation means this threshold applies only to a subset of positions in practice. UMI-based pipelines have different effective thresholds.
Q: Where can I find documentation about the mutational signature report produced by DRAGEN v4.5?
A: Detailed documentation about mutational signature / biomarker report produced by DRAGEN v4.5 can be found here: https://help.dragen.illumina.com/dragen-v4.5/product-guides/dragen-v4.5/dragen-dna-pipeline/biomarkers/biomarker-mutational-signatures
Q: Does DRAGEN v4.5 support detection of structural variants and gene fusions, and in which workflows?
A: Yes. Gene fusions are detected from RNA in the TSO500 workflow (e.g., MET, EGFR, AR in solid tissue) and in TSO500 ctDNA. DNA-based SV calling supports fusion/translocation detection in somatic panels and WGS applications (tumor-normal solid, tumor-only heme). Additional fusion support for TSO500 DNA solid tumor workflows is planned. The DRAGEN Recipes are the recommended starting point.
Q: Does DRAGEN v4.5 support the pangenome reference for the somatic workflow?
A: The somatic pipeline currently supports the pangenome reference. Formal recommendation will follow validation completion in the next release.

Q: Does the ICI support the integrated reporting of somatic, germline, and RNA findings ?
A: Yes. ICI supports combined reporting of somatic, germline findings from both DNA and RNA, with information configurable from third-party or other knowledgebases. Upcoming releases this year will add DRAGEN germline pipeline support, ACMG classification for germline variants, and expanded OncoKB germline evidence.
Q: Where can I get the best practices for germline-matched tumor analysis (SBS96, TMB, etc.)? Does Nirvana support VCF output?
A: Best practices can be found in the DRAGEN Recipes: https://help.dragen.illumina.com/dragen-v4.5/product-guides/dragen-v4.5/dragen-recipes . Nirvana supports VCF output for a subset of annotations: https://help.connected.illumina.com/annotation/v3.27/file-formats/illumina-annotator-vcf-file-format
Q: Are there known discordances between PiVAT (Pillar) and DRAGEN on amplicon panels?
A: No known discordances with DRAGEN v4.5 on currently advertised panels .
Q: Is benchmarking data available for the Pillar OncoReveal panel?
A: Yes, benchmarking data for the Pillar OncoReveal panel is available and will be included in an upcoming Amplicon application note covering Pillar panels to be released in early July. The benchmarking was performed using a combination of well-characterized reference materials, including publicly available samples (e.g., SeraCare, Horizon Discovery, and cell lines), along with internal control samples.
04 — DRAGEN TruPath Genome
Q: Is TruPath Genome part of DRAGEN v4.5 software?
A: Yes. DRAGEN supports the full TruPath Genome analysis workflow.
Q: Are there plans to run TruPath Genome on NovaSeq X?
A: Not at this time. Processing runs in the cloud or on-premises to avoid blocking sequencer throughput.
Q: Is short-read variant phasing a new capability in DRAGEN v4.5?
A: Phasing is new in the TruPath Genome workflow but is not part of the standard DRAGEN small variant pipeline.
05 — DRAGEN Multiomics
Q: In the 5-base methylation pipeline, does SV calling integrate methylation data to flag SVs that disrupt regulatory methylation patterns?
A: The SV calling algorithm has been updated to accommodate methylation conversions, but methylation-aware SV reporting is not yet implemented. This is a planned objective for a future release.
Q: Do the new v4.5 features apply to the somatic enrichment 5-base pipeline?
A: Yes. Methylation reporting at small variants (including allele-specific methylation), CNV detection, and SV detection are all enabled for 5-base enrichment pipeline.

Q: If samples were analyzed with DRAGEN v4.4.6, is a full rerun recommended?
A: Rerunning from FASTQ is preferred to ensure consistency between the mapper and variant caller versions.
Q: Which pipeline is recommended for SNP calling from RNA-seq data?
A: The DRAGEN RNA small variant caller is documented here: https://help.dragen.illumina.com/dragen-v4.5/product-guides/dragen-v4.5/dragen-rna-pipeline/rna-variant-calling. Downstream annotation with Illumina Connected Annotations is recommended.
06 — Significant Toolkit and Platform Updates
Q: How frequently are the annotation databases updated?
A: In ICA, DRAGEN pipelines automatically download annotation databases. For manual setup, see:
https://help.connected.illumina.com/annotation/v3.27/introduction/getting-started-dragen#quick-start-reference .
Q: Has the Ensembl annotation version in Nirvana been updated?
A: Yes, it has been updated to 115. Documentation has not been updated, but the version is visible when running the data manager.
Q: Does DRAGEN support large-scale joint variant calling (e.g., >1,000 samples)?
A: Yes, via the IGG workflow on ICA. Cohorts from 10K to 1M samples have been jointly analyzed at scale.
Q: Is shotgun metagenomics supported, and is a fully native DRAGEN metagenomics pipeline planned?
A: Shotgun metagenomics is supported via the DRAGEN Metagenomics Pipeline BaseSpace app (https://www.illumina.com/products/by-type/informatics-products/basespace-sequence-hub/apps/dragen-metagenomics-pipeline.html), which uses the DRAGEN aligner for dehosting and Kraken2 for classification. A fully DRAGEN-native metagenomics is not currently on the roadmap.
Q: Can the 16S rRNA v3–v4 pipeline achieve species-level identification?
A: I Species-level identification is possible but genus-level resolution is more reliable with v3–v4 primers. Using 2×500 bp reads with v1–v6 primers substantially improves species-level resolution.
Q: What are the runtimes for bacterial read dehosting?
A: Runtime depends on the sample composition and size . For a sample with ~98% human reads, at 40 CPUs, throughput is ~15 Gb/h when writing category-specific FASTQs. If only compositional assessment is needed (no FASTQ output), the throughput increases to~150 Gb/h.
Additional Resources
For more detailed information, please refer to:
- DRAGEN v4.5 Webinar On-Demand
- DRAGEN v4.5 User Guide
- DRAGEN v4.5 Technical Blog
- Request a Free Trial of DRAGEN v4.5
If you have additional questions, please contact your Illumina representative.
For Research Use Only. Not for use in diagnostic procedures.
M-GL-04456