- Home
- News & Updates
- DRAGEN™ v4.0 - Detecting repeat expansion variants for rare genetic disease insight
-
DRAGEN
-
Product updates
- 10/11/2022
DRAGEN™ v4.0 - Detecting repeat expansion variants for rare genetic disease insight
Short Tandem Repeat (STR) expansions are a pattern in DNA when one or more nucleotides are repeated adjacent to each other. STRs are ubiquitous throughout the human genome, with a phenotypically normal genome containing upwards of half a million STRs, usually occurring in trimers. STRs, when present in abnormal numbers, are also a major cause of over 20 severe neurological disorders. For example, C9orf72 repeat expansion causes 10% of ALS cases. While less than 30 copies (180bp) of C9orf72 is considered normal, pathogenic repeats associated with ALS have more than 30 copies and can span over 10 Kb.
C9orf72 repeat expansion causes 10% of ALS cases:
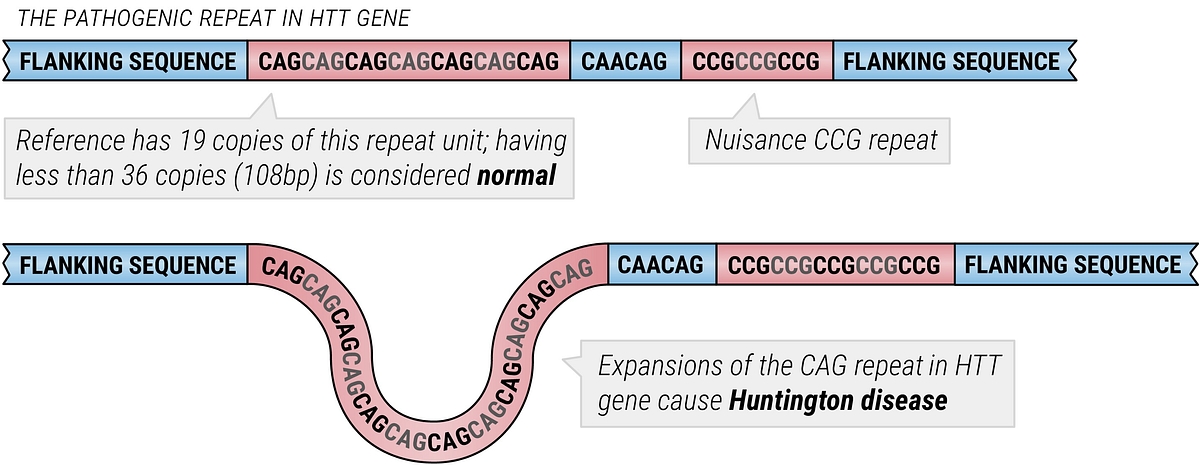
Whole genome sequencing (WGS) can be a cost-effective way to detect pathogenic repeat expansion variants and power critical insights in a variety of rare genetic diseases, including Muscular Dystrophy and Huntington’s Disease. With DRAGEN secondary analysis, we have been continuously working to maximize the value of whole genome sequencing, enabling you to capture insights for a fully featured genome.
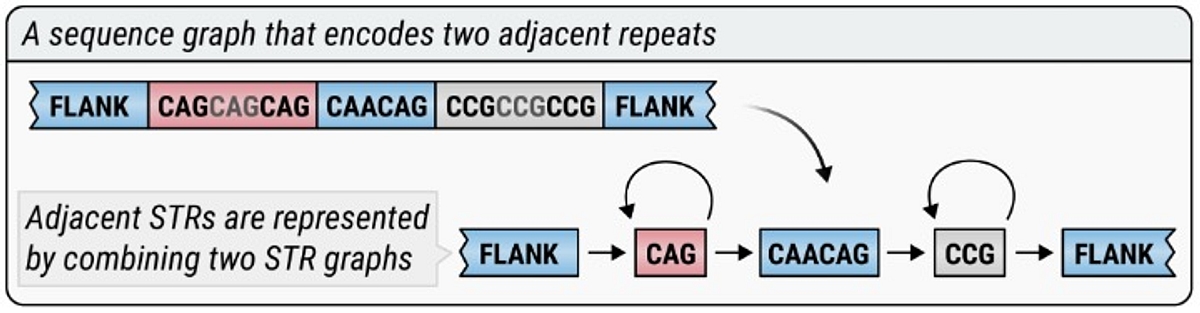
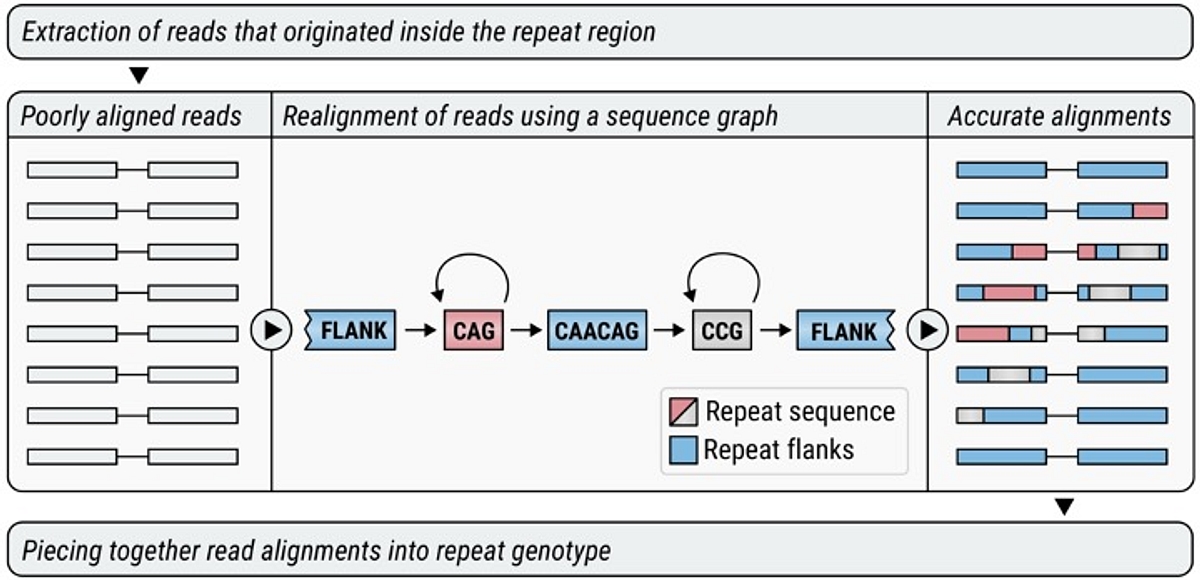
DRAGEN first introduced its ExpansionHunter tool in 2017, enabling detection of repeat-expansions and estimating repeat size to power exciting genetic disease insights. DRAGEN’s ExpansionHunter release marked the first computational method capable of detecting repeat expansions from “short read” NGS data. In 2019, we released ExpansionHunter v4 which brought genotyping of complex repeats, such as the CAG repeat flanked by a CCG repeat in the HTT gene that causes HD, using sequence graphs. The introduction of sequence graphs offered a convenient conceptual & computational model for capturing both local and genome-scale variation. ExpansionHunter performs sequence-graph based realignment of reads that originate inside and around each target repeat. It then genotypes the length of the repeat in each allele based on these graph alignments. The tool aims to estimate sizes of such repeats by performing a targeted search through a BAM/CRAM file for reads that span, flank, and are fully contained in each repeat.
ExpansionHunter uses sequence graphs to analyze repeats:
DRAGEN v4.0 brings ExpansionHunter v5, our most powerful repeat-expansion detection tool yet. ExpansionHunter v5 introduces:
- Runtime and memory usage improvements that enable scaling to large STR catalogs, building on previous releases to power exciting rare genetic disease insights.
- Genotyping of 2x the number of loci compared to ExpansionHunter v4
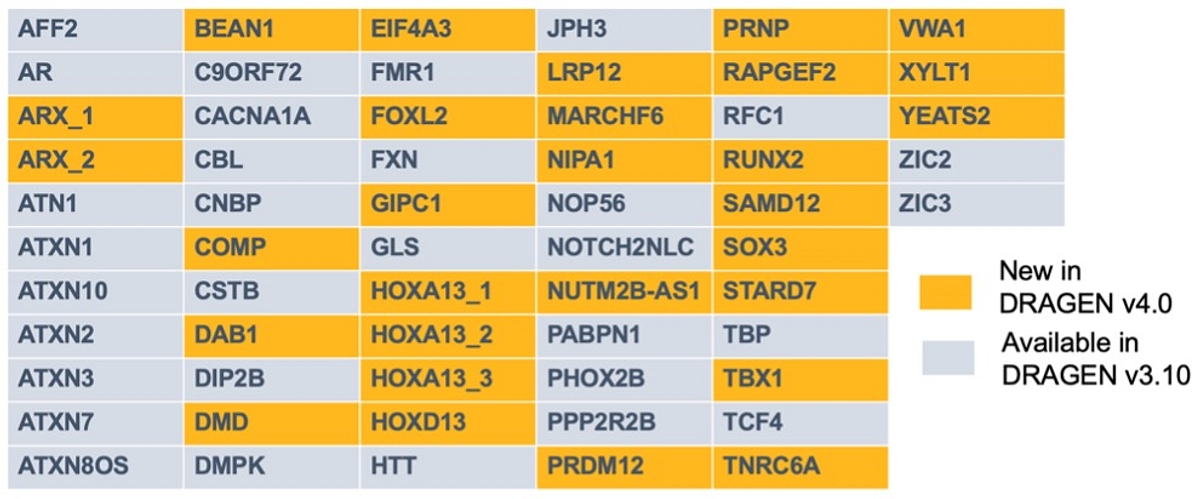
- Expanded default catalog from 30 loci to 60 pathogenic STR loci (including 30 from gnomAD)
- Optional ability to call repeats on an expanded catalog of ~50k polymorphic STR loci with minor runtime impact
With ExpansionHunter v5, you can now uncover powerful insights for rare genetic diseases like Spinocerebellar Ataxia 31, which is potentially caused by mutations in challenging genes like BEAN1.
The ExpansionHunter Gene Catalog:
With other variant callers, ExpansionHunter adds negligible overhead with default catalog and ~8-10 min overhead with expanded catalog on f1.4xlarge. Linux and macOS operating systems are currently supported.
ExpansionHunter v5 is now available on a variety of DRAGEN v4.0 deployments. These include:
- ExpansionHunter application on BaseSpace™ Sequence Hub
- Illumina Connected Analytics
- DRAGEN on multi-cloud BYOL (AWS, Azure)
Learn More:
The DRAGEN 4.0 release is packed full of comprehensiveness, accuracy, and efficiency gains, and this blog has only touched the surface.
For an in-depth summary of all DRAGEN 4.0 features, view our user guide.
Visit the DRAGEN page to learn more about our technology and other updates, including our latest v4 server release.
Other Resources:
For Research Use Only. Not for use in diagnostic procedures
M-GL-01228