- Home
- News & Updates
- Your PIPseq workflow is consolidating into DRAGEN
-
DRAGEN
-
News
- 07/15/2026
Your PIPseq workflow is consolidating into DRAGEN
Here's what to expect.
DRAGEN™ v4.5 introduces the DRAGEN scRNA with STAR (Spliced Transcripts Alignment to a Reference) pipeline, which brings the STAR aligner into the DRAGEN single-cell RNA-seq pipeline. Researchers who rely on STAR-based workflows or need continuity with STAR-aligned reference datasets can now run STAR-based analyses without leaving DRAGEN. This means you can choose either the DRAGEN aligner or the STAR aligner within DRAGEN. When STAR is enabled, only the alignment step changes; read preprocessing, barcode handling, UMI collapsing, MAPQ-based filtering, and quantification all remain in DRAGEN.
As part of this update, we will also sunset the legacy Fluent software, PIPseeker, in favor of the DRAGEN-based approach. Just as important, the DRAGEN scRNA with STAR pipeline is not simply PIPseeker moved into DRAGEN. PIPseeker uses STAR with its own chemistry-specific preprocessing, barcode calling, and MAPQ handling, while the DRAGEN scRNA with STAR pipeline uses STAR only for alignment and relies on DRAGEN for the rest of the workflow.
So, what should you expect when moving from the native DRAGEN research workflow or PIPseeker to the DRAGEN scRNA with STAR pipeline? Three STAR-related behaviors shape the answer: trimming, splice junction detection, and multimapping.
Two changes are coming to how you analyze PIPseq single-cell RNA data:
- DRAGEN v4.5 now supports STAR as an alignment option within the DRAGEN scRNA with STAR pipeline, giving you a DRAGEN-only path for scRNA analysis.
- PIPseeker is being sunset in favor of the DRAGEN scRNA with STAR pipeline.
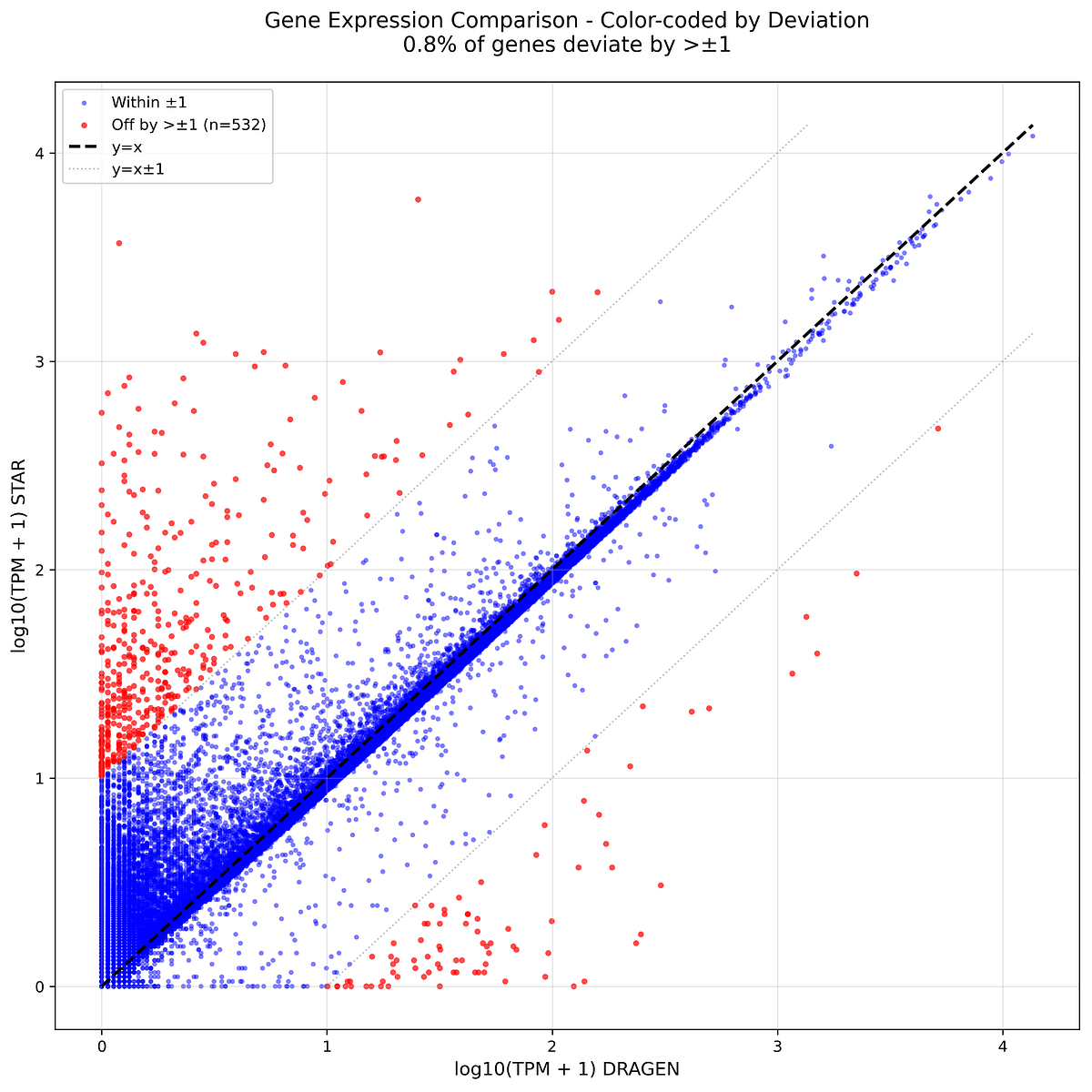
Most genes fall close to the diagonal, indicating strong overall concordance between the DRAGEN scRNA with STAR pipeline and the native DRAGEN workflow. The largest outliers are concentrated in pseudogenes and lncRNAs, consistent with differences in multimapping behavior.
Whether you're coming from PIPseeker or the native DRAGEN workflow, your analysis now runs in one supported environment. When STAR is enabled, only the alignment step changes: read preprocessing, cell barcode handling, molecular counting, MAPQ-based filtering, and quantification all remain in DRAGEN.
Before implementing this change in DRAGEN v4.5, we tested the DRAGEN scRNA with STAR pipeline and compared results from both the native DRAGEN research workflow and PIPseeker. This post explains the findings of our comparison and outlines what stays the same, what changes, and what settings to use.
What stays the same
Before getting into differences, the important context is that the core biology is preserved. In our comparison, the strongest agreement was at the cell level, and most of the remaining gene-level disagreement was concentrated in low-expression and hard-to-map gene classes rather than across the broader transcriptome. In our comparison, we found:
- Cell-level correlation between the DRAGEN scRNA with STAR pipeline (with the recommended settings outlined later in this blog) and the native DRAGEN research workflow was 0.9983.
- The core protein-coding transcriptome remained highly concordant: most genes fall close to the diagonal in gene-level comparisons, while the largest outliers are concentrated in pseudogenes, lncRNAs, and other repetitive loci.
- The differences that do exist are concentrated in low-expression genes, pseudogenes, lncRNAs, and splice junction behavior, not in the genes that typically drive biological interpretation. In this comparison, lncRNAs and pseudogenes accounted for about 81% of the genes detected only by the DRAGEN scRNA with STAR pipeline (using the recommended settings).
- Barcode processing, molecular counting, MAPQ filtering, and quantification are identical regardless of which aligner you choose. These steps remain in DRAGEN; only the alignment step changes.
Three workflows in context
Here's the simplest way to think about the three workflows discussed in this post.
- Native DRAGEN workflow: DRAGEN aligner + DRAGEN processing
- DRAGEN scRNA with STAR pipeline: STAR aligner + DRAGEN processing
- PIPseeker: STAR aligner + its own preprocessing and quantification in the legacy Fluent software
In our comparison, the native DRAGEN research workflow completed about 4× faster than the DRAGEN scRNA with STAR pipeline. Based on our comparison, the runtime difference reflects more than the aligner alone: the native DRAGEN research workflow integrates preprocessing and filtering into a single end-to-end research workflow, while the DRAGEN scRNA with STAR pipeline processes more reads downstream because the native DRAGEN research workflow would trim or discard many of them earlier.
The DRAGEN scRNA with STAR pipeline is not PIPseeker moved to DRAGEN. PIPseeker uses its own chemistry-specific preprocessing, barcode calling, quantification, and barcode allowlist, so its cell barcodes do not directly overlap with the DRAGEN call set. In contrast, the DRAGEN scRNA with STAR pipeline uses STAR only for alignment and relies on DRAGEN for the rest of the workflow. In other words, this comparison isolates the effect of changing the aligner, while PIPseeker serves as a STAR-based reference point rather than a one-to-one implementation match.
What changes when you use STAR
When STAR handles alignment inside DRAGEN for PIPseq data, three behaviors shape the output.
1) Trimming: more reads enter alignment
In our comparison, the native DRAGEN research workflow applied extensive preprocessing before alignment, including a fixed 1 bp 5' trim, removal of 5' TSO and SMART PCR primer sequence when present, 3' polyA removal, 3' poly-G removal, and filtering out reads shorter than 20 bp after trimming. On our test dataset, it trimmed an average of 21 bases per read and filtered 12.11% of reads (14.9M reads) for insufficient length.
In our comparison, STAR showed limited built-in trimming and could not natively detect and remove variable-length 5' adapter sequences such as TSO or SMART PCR primer sequence. Without adjustment, the DRAGEN scRNA with STAR pipeline processed 11.2% more input reads than did the native DRAGEN research workflow (120.2M vs. 108.1M). Many of the additional genes detected by only the DRAGEN scRNA with STAR pipeline (6,575 genes) showed very low UMI support: the median total UMI count was 2, nearly 75% had 5 or fewer total UMIs, and 40% had exactly 1 UMI. In this comparison, these extra genes were enriched for pseudogenes and lncRNAs rather than protein-coding genes, consistent with low-confidence or ambiguous signal rather than robust biological expression.
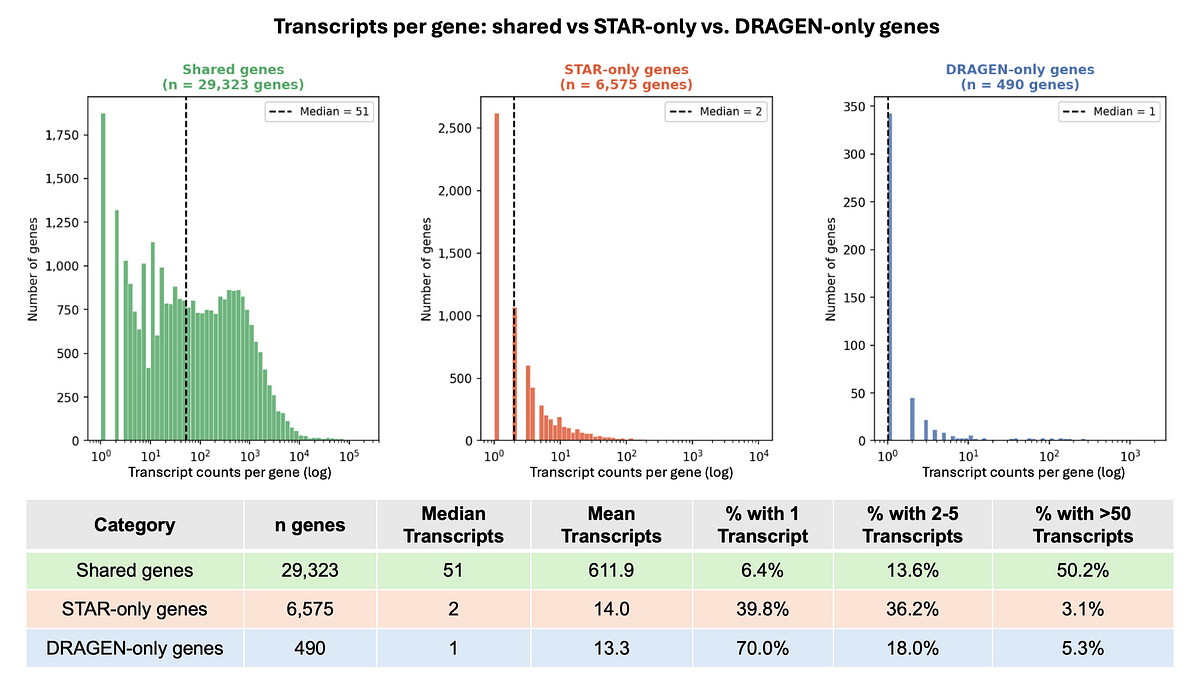
Genes detected only in the DRAGEN scRNA with STAR pipeline cluster at very low UMI counts, supporting our conclusion that, in this comparison, many of these additional calls reflect ambiguous or low-confidence signals rather than robust biological expression.
2) Splice junctions: STAR's default intron limit is wider
In our results, STAR defaulted to a maximum intron length of 1 million bp, while the native DRAGEN research workflow limited intron detection to 50 kb. In practice, the default DRAGEN scRNA with STAR pipeline reported nearly twice as many unique splice junctions (128,775 vs. 65,783), with the excess concentrated at long intron lengths and largely unannotated. Setting the alignIntronMax to 25000 brought this down to 100,043 junctions. This suppressed the most artifact-prone long-range calls, but not all residual differences, because the two aligners still use fundamentally different splice-junction detection algorithms. If your research depends on splice-junction interpretation, review long-intron junction calls with extra scrutiny.
3) Multimapping: the aligner scores ambiguous reads differently
This is the most fundamental remaining difference between the aligners, and it cannot be fully eliminated through settings.
In our results, STAR assigned MAPQ=3 to reads with two equally good alignments, allowing them to pass through the DRAGEN MAPQ≥3 filter and potentially be counted at two loci. The native DRAGEN research workflow assigned MAPQ=0 to those same reads and discarded them. We found that this translated into roughly 7.3M reads dropped by the MAPQ filter using the DRAGEN scRNA with STAR pipeline (recommended settings) versus about 16.6M in native DRAGEN. As a result, in our comparison, STAR-based runs sometimes assigned counts where the native DRAGEN research workflow did not. The native DRAGEN research workflow also retained signal differently at certain repetitive loci, such as HLA, because the aligners resolved ambiguity differently.
Additionally, this pattern was most visible in pseudogenes and lncRNAs, where STAR-based runs tended to produce higher counts, and in HLA loci, where native DRAGEN tended to produce higher counts.
Most genes fall close to the diagonal, indicating strong overall concordance between the DRAGEN scRNA with STAR pipeline and the native DRAGEN workflow. The largest outliers are concentrated in pseudogenes and lncRNAs, consistent with differences in multimapping behavior.
Where the differences matter most
Taken together, these results show that most of the difference between the DRAGEN scRNA with STAR pipeline and the native DRAGEN research workflow comes from preprocessing, splice junction detection, and multimapper scoring. At the read level, the two recommended-setting configurations agreed on gene assignment for about 81.5% of reads, and the remaining disagreement was driven most often by one workflow assigning a gene where the other left the read unassigned.
Our comparison showed:
- Higher counts in the STAR-based workflows often reflected a combination of
- Minimal trimming
- Permissive intron detection
- Multimapping read inclusion
- The native DRAGEN research workflow generally produced
- Fewer counts at ambiguous loci (more conservative)
- Stronger suppression of low-confidence multimapper signal
Importantly, the core protein-coding transcriptome remained highly concordant in our comparison. The biggest differences were concentrated in low-expression genes, pseudogenes, lncRNAs, highly repetitive regions, and splice-junction behavior.
How to bring the DRAGEN scRNA with STAR pipeline closer to the native DRAGEN research workflow
If you plan to use the DRAGEN scRNA with STAR pipeline for PIPseq data, these are the most important settings:
- alignIntronMax 25000
- clip5pNbases 1
- clip3pAdapterSeq polyA
Recommended settings for the DRAGEN scRNA with STAR pipeline
If you're using the DRAGEN scRNA with STAR pipeline for PIPseq data you can either trim the reads using Cutadapt with these instructions or the following settings:

We found these settings improved cell-level correlation from 0.9935 to 0.9983 and brought the STAR read input count into line with native DRAGEN by combining a fixed 5' base clip with polyA trimming. They do not fully replicate the native DRAGEN research workflow's dynamic 5' adapter detection or change STAR's multimapper scoring, so some gene-level differences remain, particularly at pseudogenes, lncRNAs, HLA, and other highly repetitive loci.
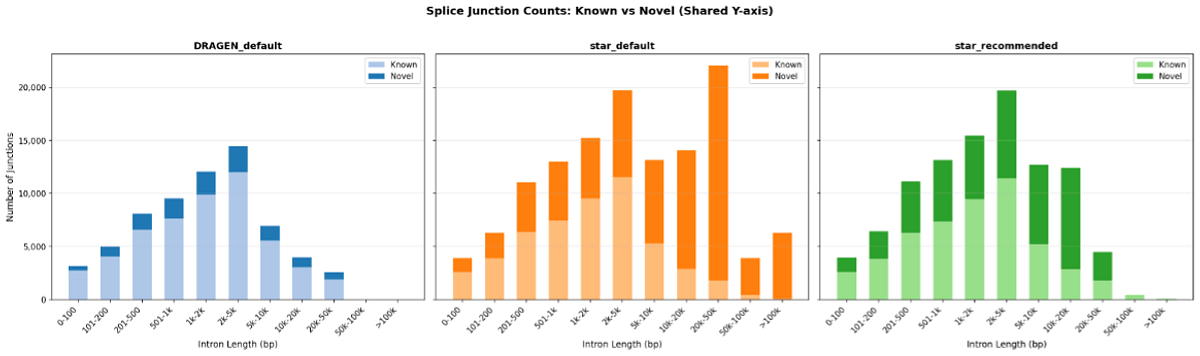
Comparing the native DRAGEN research workflow to the DRAGEN scRNA with STAR pipeline, the recommended STAR settings substantially suppress long-range novel splice junctions and bring junction profiles closer to native DRAGEN results, though some aligner-specific differences remain.
Applying these settings improved our cell-level correlation with the native DRAGEN research workflow from 0.9935 to 0.9983, but some gene-level differences remained because multimapper handling is intrinsic to the aligner rather than fully parameter-tunable.
When to use each approach?
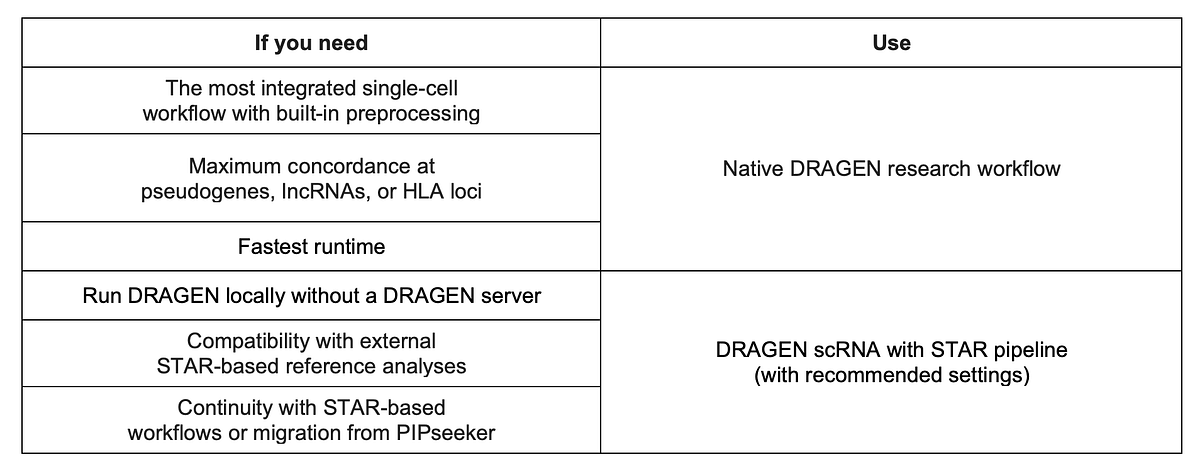
What you need to do
If you're a PIPseeker user:
- Your analysis path is moving to the DRAGEN scRNA with STAR pipeline. Apply the three recommended settings above for the closest match to your previous workflow.
- Your past PIPseeker results remain valid. The differences described here reflect aligner-level behaviors, not errors in previous analyses.
- Expect differences in pseudogene handling. In our comparison, PIPseeker suppressed the mitochondrial pseudogene class entirely, while the native DRAGEN and DRAGEN scRNA with STAR pipelines retained pseudogene signal in the output, with STAR-based runs showing the highest counts at these repetitive loci.
- For step-by-step guidance on transitioning, see the user guide.
If you're a native DRAGEN user:
- Your default workflow is unchanged. The DRAGEN scRNA with STAR pipeline is an additional option, not a replacement.
- If you choose to use the DRAGEN scRNA with STAR pipeline for compatibility reasons, apply the recommended settings. They improve cell-level correlation from 0.9935 to 0.9983, but they do not fully reproduce the native DRAGEN workflow's trimming or multimapper behavior.
For all users:
- Our comparison shows that the core protein-coding transcriptome remains highly concordant across all three conditions, while the largest residual differences are concentrated in low-expression genes, pseudogenes, lncRNAs, HLA, and splice-junction behavior.
- If your study depends on low-expression genes, pseudogenes, lncRNAs, HLA, or splice junction interpretation, review these comparison findings carefully, because those are the areas where aligner-specific differences continue to matter most.
Key takeaway
If you need compatibility with STAR-based workflows, the DRAGEN scRNA with STAR pipeline (using recommended settings) offers the closest approximation to the native DRAGEN research workflow within the DRAGEN environment. In our comparison, those settings reduced the biggest trimming- and intron-related discrepancies and improved cell-level correlation to 0.9983.
If your analysis depends on strict concordance, low-expression genes, pseudogenes, lncRNAs, HLA, or splice-junction interpretation, the native DRAGEN research workflow remains the most direct choice because those are the areas where aligner-specific trimming, junction calling, and multimapper scoring differences continue to matter most.
If speed is your priority, the native DRAGEN research workflow performed about 4× faster than the STAR-based path in our comparison.
---
For Research Use Only. Not for use in diagnostic procedures.
M-GL-04595